
![]()
![]()
![]()
![]()
![]()
![]()
|
|
| [an error occurred while processing this directive] |
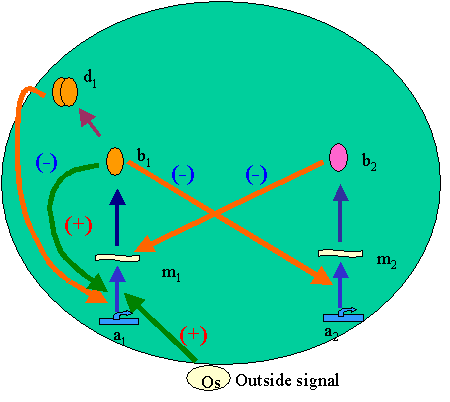
Principles of mathematical modelling (demonstration model)A particular example illustrates best the procedure of constructing the gene network model. Let us consider a fragment of gene network (Fig. 2) describing the system switching over from one mode into the other depending on an outside signal (Os), representing a model of abstract asymmetrical molecular trigger.
The system includes two genes, a1 and a2, coding for individual mRNAs, m1 and m2, wherefrom proteins b1 and b2 are translated. Monomer of protein b1 forms dimer d1. Protein b2 is a specific factor responsible for degrading mRNA m1; protein b1 inhibits the transcription of gene a2 and simultaneously activates transcription of its own gene a1; while dimer d1 of protein b1 inhibits transcription from its own gene a1. In norm, gene a1 is inactive. Its primary activation depends on the outside signal Os. Gene a2 in norm synthesizes mRNA with a certain nonzero activity. Both mRNA and proteins have limited life spans. Let us enumerate all the elementary processes specified for this network:
Fig. 3 shows the formal description of all the elementary processes of the gene network.
The description complies with the following scheme. Lines marked with identifiers /cc/, /fb/, /xx/, and /ap/ (more than one lines /cc/ and /ap/ are allowed) contain the information on the process. Certain processes may be described in combination. In our example, these are elementary processes 2 and 3, describing mRNA translation; processes 10 and 11; and processes 12 and 13. Line /cc/ contains comments clarifying briefly the essence of a process. For example, process 1 describes constitutive transcription of gene a2, a pair of processes 2 and 3 describes translation of mRNAs synthesized from genes a1 and a2, etc. Line /fb/ indicates the number of a formal block as assigned in Fig. 1. This number describes completely the formalism accepted for description of the process. For example, formal block 4 (generalized Michaelis- Menten scheme 1) is used for describing processes 1- 5 and 9; block 1 (bimolecular reaction), for processes 6- 7; or block 2 (irreversible monomolecular reaction), for processes 10- 13. Next comes line /xx/, describing actual names ascribed to the variables of a block while describing a particular process. For the sake of definiteness, we will call these named variable as products. Within each list, products are separated with commas. The order of products conforms strictly to the order of formal variables in the corresponding blocks listed in Fig. 1. Description of a process is concluded with one or several lines /ap/ containing two types of information. The first information block (detailed below) is located before a colon (:), having a meaning of separation. The second information block located after the colon contains the data on constants in a form of list of actual parameter names and their values separated with commas. Name of a parameter and its value is separated with equal sign (=). Let us term a parameter with assigned name as constant. Name of the product (constant) is specified in a binary form as name{pseudoextension}. In general case, any combinations (including empty) of letters, digits, and other symbols may be used as names (pseudoextensions). The first part, name, is the name of the corresponding product (constant). In the current example, gene, RNA, mRNA, ribo, protein, protein2, rgene, and signal in the lists of products and k1, k2, k3, … in the list of constants are names. Designations {}, {pol}, some}, {a1}, {a2}, and {out} are pseudoextensions of the names of corresponding products (constants). Pseudoextension {} is considered empty. Braces are symbols separating pseudoextensions. Pseudoextensions have a service meaning and are used at the second stage while constructing a particular model for correct completing of product (constant) name definitions with specialized names termed extensions. Running ahead, note that only the pair name{pseudoextension} characterizes a product (constant) integrally. However, the pair name{pseudoextension} has also a specific biological meaning. The product gene{} signifies an abstract gene or product mRNA{}, an abstract mRNA, whereas the product gene{a1} denotes particular gene a1 or RNA{pol}, particular RNA polymerase, etc. Thus, a product is characterized precisely while describing any process if, and only if its name is supplemented with nonempty pseudoextension (RNA{pol}, Ribo{some}, etc.). If the product name is supplemented with an empty pseudoextension, the product is specified only by its type (gene{}, mRNA{}, or protein{}) to be thoroughly characterized directly while constructing a particular model. Let us consider next the actual construction of a model from the processes shown in Fig. 3. For this purpose, we need to specify the housekeeping information in a form of name list, which we call MAP. The MAP list in the general GCKSM version is a linearly ordered oriented list of names. Genetic maps, where genes are oriented and linearly ordered, represent most close analogy to the MAP list. Physical carriers of genetic maps—DNA or RNA—can be circular, linear, or constituted of several physically independent molecules; therefore, MAP as a formal construction in GCKSM is endowed with the same features. Let us term the names from MAP as modules and their totality as structure. Here, we are describing the simplest GCKSM version, where the structure is considered as an unordered unoriented list of names. This version is sufficient to construct a wide class of models (including those described below) wherein the elementary subsystems interact mainly due to trans-effects.
Returning to the model (Fig. 4), note that description of each process contains one or several lines /ap/. Meaning of the information located after the colon is described above (constants and their values). Now we shall clarify the meaning of information located within braces before the colon. Line /ap/ may contain either one name (processes 1- 9) or more than one. In case there are several names, they are separated with commas (processes 10- 11 and 12- 13). These names are the names of modules whereof the structure is constructed. According to GCKSM, each elementary process is ascribed to a particular module. In other words, the term module is considered to mean an aggregate of elementary processes. In the model considered, all the elementary processes are grouped into five modules {a1}, {a2}, {ma1}, {ma1a2} and {ma2a1}. For example, module {a1} is a set of elementary processes shown in Fig. 4. Elementary processes are arranged as follows. All the processes containing module {a1} in line /ap/ are sorted out. The list of constants contained in the same line as the name of the module in question is associated with each process. In each process, all the empty pseudoextensions of products and constants are substituted with {a1}. Consequently, we obtain a set of elementary processes assigned to the module a1. Similarly, the rest modules are filled. Thus, module a2 is composed of elementary processes 1, 3, 11, and 13, while module {ma1} is represented solely by elementary process 5; {ma1a2}, by process 9; and {ma2a1}, by process 6. Modules are construction bricks of GCKSM. Only 5 modules allow 31 structures to be constructed: 5 one-element; 10 two-element, 10 three element, 5 four-element, and 1 five-element (module repeats are not considered as in this module they do not have any independent sense). Let us consider several structures. The five-element structure ĚŔĐ = ({a1},{ŕ2},{ma1},{ma2a1},{ma1a2}) presents the model of initial gene network. The structure MAP = ({a2}) simulates the gene network fragment that includes gene a2, mRNA m2, and protein b2; ĚŔĐ = ({ŕ2},{ma1a2}), the same fragment of the gene network supplemented with the inhibition function of protein b2 (Fig. 2). The structure ĚŔĐ = ({ŕ1}) allows the gene network fragment comprising gene a1, mRNA m1, protein b1, its dimer d1, and outside signal Os to be constructed (Fig. 2). Let us see now how a model is constructed according to the structure specified commencing from the structure ĚŔĐ1 =({ŕ1},{ma1},{ma1a2}). We have already traced out all the elementary processes of the module {a1} (Fig. 4) and will now add to them all the elementary processes of the module {ma1}. Recall that for this purpose we need to take from the model (Fig. 3) all the typical processes ascribed to the module {ma1} (in this case, it is process 8 in Fig. 3), substitute all the empty pseudoextensions with pseudoextensions {ma1}, and add the resulting elementary processes to the set of module {ŕ1} processes (Fig. 4). Similarly, the elementary processes of the module {ma1a2} are added to the set of processes constituting the essence of module {ŕ1},{ma1}. The resulting aggregate will represent the model in question constructed in terms of elementary processes. Let us designate this model as model(ĚŔĐ1). All the products and constants of this model are fully characterized through their names and extensions. This unambiguously relates the products and elementary processes they enter, thereby integrating these processes through identical products into a unified scheme. Since this model is constructed in terms of kinetic blocks (recall that we used formal blocks 1, 2, and 4 while describing the elementary processes), it may be rewritten as a set of common autonomous equations. For this purpose, it is necessary to apply the rule of summing the instantaneous rates of product concentration changes over all the processes wherein these products are involved. We will explain the procedure of setting up such equations by example of the product mRNA{ŕ1}. This product is involved in processes 1, 4, 5, 9, and 10 (Fig. 4). The instantaneous rate of its change in process 2 (formal block 4) equals S2 = 0; in process 4 (formal block 4) amounts to S4 = [ko3{a1} * gene{a1} * RNA{pol} * signal{out}/(km3{a1}2 + + km3{a1} * (gene{a1} + RNA{pol} + signal{out}) + gene{a1} * RNA{pol} + gene{a1} * signal{out} + RNA{pol} * signal{out})]; in process 5 (formal block 4), S5 = [ko4{ma1} * gene{a1} * RNA{pol} * protein{a1}/(km4{ma1}2 + + km4{ma1} * (gene{a1} + RNA{pol} + protein{a1}) + gene{a1} * RNA{pol} + gene{a1} * protein{a1} + RNA{pol} * protein{a1})]; in process 9 (formal block 4), S9 = [ko8{ma1a2} * mRNA{a1} * protein{a2}/(km8{ma1a2} + mRNA{a1} + protein{a})]; and in process 10 (formal block 2), S10 = - k9{a1} * mRNA{ŕ1}. Consequently, the resulting equation of the instantaneous rates of changes in the product concentration is d(mRNA{ŕ1 })/d t= S2 + S4 + S5 + S9 + S10 (all the rates are summed). Repeating similar procedures for the rest products, we obtain the description of model(ĚŔĐ1) in a differential form: d(mRNA{ŕ1})/dt = ko3{a1} * gene{a1} * RNA{pol} * signal{out}/(km3{a1}2 + + km3{a1} * (gene{a1} + RNA{pol} + signal{out}) + gene{a1} * RNA{pol} + gene{a1} * signal{out} + RNA{pol} * signal{out}) + ko4{ma1} * gene{a1} * RNA{pol} * protein{a1}/(km4{ma1}2 + + km4{ma1} * (gene{a1} + RNA{pol} + protein{a1}) + gene{a1} * RNA{pol} + gene{a1} * protein{a1} + RNA{pol} * protein{a1}) – ko8{ma1a2} * mRNA{a1} * protein{a2}/(km8{ma1a2} + mRNA{a1} + protein{a}) - k9{a1} * mRNA{ŕ1} d(protein{ŕ1})/dt = ko2{a1} * mRNA{a1} * ribo{some}/(km2{a1} + mRNA{a1} + ribo{some}) + 2 * (- k61{a1} * (protein{ŕ1})2 + + k62{a1} * protein2{ŕ1}) –k10{a1} * protein {ŕ1} d(protein2{ŕ1})/dt = k61{a1} * (protein{ŕ1})2 - k62{a1} * protein2{ŕ1} –k71{a1} * gene{a1} * protein2{a1} + k72{a1} * rgene{a1} d(gene{a1})/dt = –k71{a1} * gene{a1} * protein2{ŕ1} + k72{a1} * rgene2{a1} d(rgene2{a1})/d t= k71{a1} * gene{a1} * protein2{ŕ1} - k72{a1} * rgene2{a1} If another structure is considered, it will give rise to another model. The model of initial gene network is constructed if the structure ĚŔĐ = ({a1},{ŕ2},{ma1},{ma2a1},{ma1a2}) is specified. Thus, specifying different structures, we can construct not only one model, but also a series of models. Evidently, there is no need in constructing all the models at once. Each particular model is constructed only as needed. Note in conclusion that our method in its most general realization implies the possibility of simulation not only trans-, but also cis-interactions. The latter should be taken into account in those cases when patterns of gene network performance depend not only on the functions, but also on relative location of the genes involved.
|



{kind=link}