What
does PDBSiteScan do?
How
does PDBSiteScan work?
·
PDBSiteScan
provides automated search of
three-dimensional (3D) protein fragments similar in structure to known
functional sites. A collection of known sites we designated as PDBSite
was set up by automated processing of the PDB database, using the data on site
localization in the SITE field. Additionally, protein-protein interaction sites
were generated by analysis of contact residues in heterocomplexes.
We accepted a residue as contact, if it had at least three atoms whose distance
from any atoms of the partner chain was smaller than 5 Å.
·
The algorithm
developed is based on exhaustion of all the possible combinations of protein
positions to be compared with the site. PDBSiteScan
accomplishes the following steps. At the first step, the amino acids of a
protein part and the site are compared. If they are identical, their 3D
structures are compared at the second step. If the maximum distance mismatch
(MDM) of superimposed structures is smaller that the user-specified value, the
fragment examined is added to the list of the results. Our approach considers
only atoms N, Ca
,
C that define the
spatial orientation of residues.
Using
the PDBSiteScan Interface
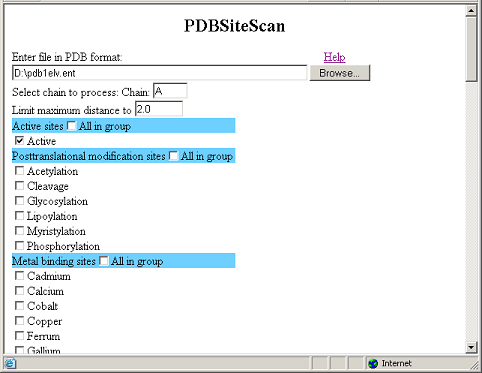
Input
for PDBSiteScan
Step
1.
Pick a structure to scan
·
Input a
filename of the tertiary structure of a protein under study into the text
window. The tertiary structure should be in the PDB format. You can upload it by
clicking the "Browse" button.
·
Input ID chain
of the protein to be analyzed.
Step2.
Specify the threshold value of the MDM
Decision making, whether
or not to include the canning results for the potential functional sites,
structurally similar to the real, in the results, depends on the MDM threshold
value. The requirements for the structural similarity of the potential and real
sites become less stringent with decreasing threshold MDM values. At high
values, potential sites very different from the real are not discarded by the
program, thereby producing overprediction of
functional sites; at low values, the potential sites even structurally similar
to the real, can be discarded thereby producing underprediction
of functional sites. The optimum MDM threshold values vary in the narrow 1.0 –
2.5 A range.
Step
3.
Choose a type of site to scan
PDBSiteScan
searches active sites, binding sites and posttranslational modification sites in
3D protein structure. To search sites of a particular type, click checkbox next
to the site name. When a site group is marked with a checkbox “All in a
group”, all the sites in the group become predictable by PDBSiteScan.
Step
4.
Submit query
Click "Scan"
button at the bottom of the page.
Step
5.
View results
·
To obtain a
structural alignment, choose the site of interest from the list of the
identified sites by placing a checkbox next to the site name. Then, click
“Download structure alignment as PDB file”.
·
To view the PDBSite
database entry, corresponding to the identified site, click ID of the given
site.
PDBSiteScan
Output
·
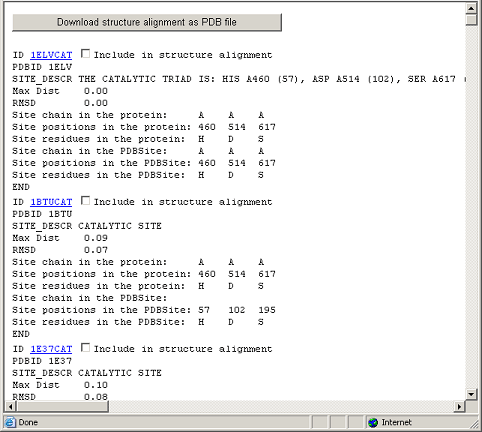
The output
contains information on the superimposition for each site–protein fragment
pair. It also includes a unique PDBSite identifier
of the site; the protein PDB ID from which the site was extracted; site
description; the MDM and Root Mean Square Deviation (RMSD) values for the
identified protein fragment; structural alignment data. Each result is linked to
the complete information on the site in PDBSite.
·
PDBSiteScan
provides structure site–protein alignment as a PDB file. This allows
visualization of the structure alignment by the popular software, such as Chime
and RasMol.
Example
Let us consider the
application of PDBSiteScan, using a
recognition of a catalytic site in the 1ELV protein (the
HYDROLASE family) as an example.
This figure demonstrates
the input data for PDBSiteScan.
This figure demonstrates the result of PDBSiteScan operation.
Comments and questions are welcome to Vladimir Ivanisenko