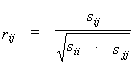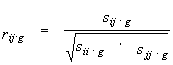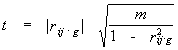|
(1) |
DETECTING DIRECT CORRELATIONS BETWEEN POSITIONS IN MULTIPLE ALIGMENT OF AMINO ACID SEQUENCES
D. A. AFONNIKOV, YU. V. KONDRAKHIN, I. I. TITOV, N. A. KOLCHANOV
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090, Russia
E-mail : ada@bionet.nsc.ru
Keywords: multiple alignment, bZIP-domain, amino acids, physico-chemical properties, substitutions, correlations.
ABSTRACT
The method is developed to detect direct correlations between positions in multiple alignment of amino acid sequences. It estimates mutual dependence of physical or chemical characteristic quantities for all the possible pairs of alignment positions based on the partial correlation coefficient as a measure of statistical linkage between them. The approach allows to estimate correlation between the pair of positions eliminating the influence of the remained sequence positions. DNA-binding regions of transcription factors of the CREB and AP-1 families were analyzed by this method. A large number of direct correlations between position pairs have been revealed. The results obtained are in good agreement with the available experimental and structural data on these transcription factors.
INTRODUCTION
One of the main tasks of genome computer analysis is to reveal regularities of structural and functional organization of proteins encoded by genes. In common practice, multiple alignment serves as a reliable tool of analysis of homologous protein sequences. Statistical dependencies between alignment positions provide an important information on possible interactions between protein residues as reflecting proteins structure and function. It should be noted that correlation analysis of multiple alignments is widespread technique considered as the most reliable to predict RNA secondary structure (Gutell et al, 1992; Chiu and Kolodziejczak, 1991; Kolchanov et al, 1996). As for proteins, analysis of pair amino acid correlations seems a troublesome problem due to a greater number of different types of monomers and their complicated interactions. Nevertheless, a considerable number of new approaches have been suggested during the last decade to reveal correlated positions in amino acid sequence alignments. Among the techniques used to define statistical correlation between position pairs are the following: pattern matching method (Altshuh et al, 1987), informational approach (Korber et al, 1993; Clarke, 1995), analysis of position correlations on the base of different measures of amino acid relatedness (Gobel et al, 1994; Taylor and Hatrick, 1994), statistical evaluation of the pairwise amino acid substitution frequencies (Shindyalov et al, 1994), analysis of correlations in terms of physical quantities (Neher, 1994). However, a correlations thus revealed for any pair of positions might occur indirect, i.e. to be a result of interactions between these positions and certain other position(s) of the alignment. Bearing this in mind, we have developed the method that will give a chance to estimate a degree of direct correlation between every position pair and to eliminate any intermediate influence of residues in other protein positions.
METHOD
Let us consider a sample of N aligned sequences of the length L. Then we state some certain physical or chemical amino acid characteristic f (hydrophobicity, charge, side chain volume, etc.). A corresponding quantity of this characteristic could be attributed to every amino acid in alignment. In result, we obtain a matrix F whose elements Fki are quantities of f for the i-th position of the k-th sequence. This matrix considered as a set of N realizations of L-dimensional vector of real random variables f=(f1, ..., fi, ..., fL) — quantities of amino acid characteristic f at the alignment positions.
For the variable pair fi,fj their mutual distribution can be characterized by the mean values ` fi,` fj, as well as dispersions sii,sjj and covariation sij, measuring statistical dependence between these variables. Mean values might be estimated on the base of the matrix F as follows:
|
(1) |
Covariation sij (i ą j) and variance (if i=j) one can estimate by the formula
|
(2) |
For the array of L variables, their mutual distribution is described by a vector of means ` f=(` f1, ..., ` fi, ..., ` fL) and L´ L covariation matrix S=(sij). In this matrix, the elements sij are covariations of variables fi,fj for all possible position pairs. Parameters ` f and S are calculated from the matrix F on the base of (1) and (2). In case when fi values follow the Gaussian Distribution, their mutual distribution is determined completely by ` f and S (Anderson, 1958). In order to estimate the dependence between the pair of variables fi and fj there is often used an ordinary linear correlation coefficient:
|
(3) |
where sij are elements of the covariation matrix S.
However, taking into account possible intermediate interactions between the variables, this approach might lead to obtaining inadequate results. That is why we used the partial correlation coefficient as a measure of correlation between physical or chemical quantities of amino acids for the i, j pairs of alignment positions. This coefficient estimates the interaction between variables fi, and fj provided that the remained g=L-2 variables are all fixed, i.e. it allows to define a direct dependence between positions i and j of alignment. The coefficient rij.g for variable pair fi and fj is calculated from the conditional covariation matrix Sij.g as derived from matrix S by the transformation (Anderson, 1958):
Sij.g = Sij - Sij,g Sgg-1 STij,g (4)
Here Sij is a 2´ 2 submatrix of the matrix S containing the values of dispersions and covariation for the chosen positions i and j; Sgg – (L-2)´ (L-2) submatrix of dispersions and covariations for the remaining g=L-2 positions; Sij,g – 2´ (L-2) cross-covariation submatrix between variable set { i,j} and set { g} of the remaining L-2 positions; T is a transposition symbol. This transformation results in vanishing of cross-covariations between variable sets { i,j} and { g} . In this way interactions of fi,fj with the other variables could be eliminated.
The partial correlation coefficient for the pair of variables i,j is expressed through elements of the Sij.g as follows (Anderson, 1958):
|
(5) |
Under assumption of Gaussian distribution of the physical and chemical quantities throughout the independent alignment positions the following statistics
|
(6) |
has a Student's distribution (Anderson, 1958) with m = N-g-2 degrees of freedom, where N – number of sequences in the alignment. So, the correlation between pair of positions vanishes at the given level of significance if the absolute value of correlation coefficient is below critical value rc estimated by the formula (6).
This formula allows to estimate a critical value of the ordinary correlation coefficient also if to substitute the degrees of freedom by m = N-2.
It should be pointed that the distribution of amino acid physical or chemical quantities in positions of alignment differs from the Gaussian at least due to the discrete nature of quantity scales. Therefore we used an additional permutation test in order to estimate the critical value rc as irrespective to the type of potential distribution of amino acid characteristic quantities throughout the alignment positions. There were simulated 1000 samples out of the initial alignment via random amino acid permutations in each column of the alignment. In this way any potential correlation between amino acids at different positions has been eliminated with the frequencies of these amino acids unchanged. On the base of such randomized samples, one can estimate the probability prand(|rij.g|>rc) for a pair of independent positions to exceed by random the value rc of the partial correlation coefficient. The probability prand(|rij.g|>rc) was estimated as a ratio M1/M2, where M1 – number of position pairs whose value |rij.g| was greater than rc and M2 – total number of the tested pairs. The same approach has been applied to obtain prand(|rij.g|>rc) values for an ordinary correlation coefficient.
IMPLEMENTATION
System. The method described above was implemented in C on an IBM PC computer running under MS DOS operating system.
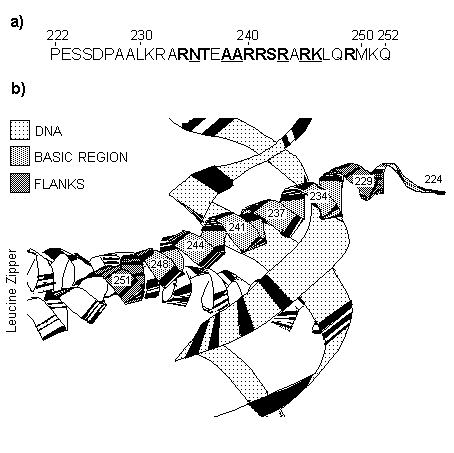
Sampling of sequences. We examined the DNA-binding regions of transcription factors belonging to the CREB and AP-1 families. These proteins binds DNA as a dimers and contains a DNA-binding domain of the bZIP class. The bZIP domain consists of the short region rich in positively charged (basic) residues at the N-end of the domain and segment containing repeated leucine residues (the leucine zipper) immediately adjacent to the C-end of the basic region (Landshultz et al, 1988). Basic region directly binds DNA and the leucine zipper serves for dimerization of proteins. Three-dimensional structure of DNA-bZIP complex has been determined (Ellenberger et al, 1992) and it is showed in Figure 1 for GCN4 protein. The basic region acts as a continuous a -helix and passes through the major groove of the DNA-binding site (Figure 1a).
For our analysis, we have chosen 49 transcription factors from the SWISS-PROT databank (Bairoch and Boeckman, 1991) listed in Appendix 1. Positioning of the DNA-binding domains in proteins was defined as being assigned in the databank. Together with the nearby regions (30 positions from the N-end and 10 positions from the C-end) the domain sequences were aligned by the multiple alignment program (Seledtsov et al, 1995). Then we considered only the DNA-binding regions with a few flanking positions at both ends (Figure 1b). There were eliminated low-variable positions with no more than two different amino acids in the alignment and positions that contain deletions. As a result, for the sample of 49 sequences 25 positions of region under consideration were taken into account.
Physical and chemical properties of amino acids. Position correlations have been considered for such amino acid characteristics as side chain volume (Chothia, 1984), hydrophobicity (Eisenberg et al, 1984) and isoelectric point (White et al, 1978).
Estimation of reliability level. The critical value rc for N=49, g=23 and reliability P=0.99 was estimated by the formula (6) as being about 0.36 for ordinary and 0.49 for partial correlation coefficients.
RESULTS
We have carried out a randomization test for the sequence sample of interest as described above. As a result, for the isoelectric point quantities and ordinary correlation coefficient we obtained prand(|rij|>0.36)» 0.011, and for partial correlation coefficient prand(|rij.g|>0.49)» 0.011. Estimations of prand for the remaining physical quantities in point have also occurred close to these values (data not given). Thus, the values of 1- prand obtained are close to the reliability level P used above to calculate rc values by the expression (6).
Calculations of the ordinary and partial correlation coefficients were obtained as symmetrical matrices whose diagram representation is given in Figure 3. The data given here might be of interest from the following viewpoints. First of all, there is a good agreement between these results and available data on the structure and function of sequences examined. Secondly, there is a striking differences between results obtained by partial and ordinary correlation coefficients.
It should be pointed firstly that the considered amino acid characteristics reflect physical inter-residue interactions as well as interactions between residues and protein environment. For the side chain volume these are sterical short-distance interactions. Isoelectric point could be associated with charge properties of amino acids. Hydrophobicity characterizes interaction of residues with aqueous solution. The last two properties for charged residues could be considered as related. One can see that matrices of the partial correlation coefficients are quite similar as obtained for hydrophobicity and isoelectric point, supposingly, due to a close nature of these properties.
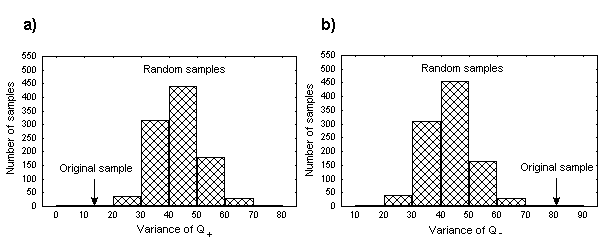
Next, there should be mentioned here a strong correlation in terms of hydrophobicity and isoelectric point quantities between positions flanking the DNA-binding region. The latter could be detected by the blocks of significant correlation coefficients in the left bottom parts of the respective matrices. For example, for the isoelectric point - they are positions 227-230 and 249-252 that form a complete helix turns at the N- and C-ends, respectively. They are designated as “flanks” in Figure 1b. Analysis of isoelectric point quantities for these region has evidenced some other interesting features. The total sum Q + =QN+QC of this quantities in mentioned helical regions has occurred conservative for the proteins of interest (QN and QC denotes summary isoelectric point quantities at positions 227-230 and 249-252 respectively). One can see that Q+ variation calculated for real proteins is significantly lower than that calculated for random samples(Figure 3a). It should be underlined here that QN and QC values are reflecting a summary charge at the N- and C-ends of corresponding helices. Thus, the obtained results could indicate that the summary charge of two mentioned regions in sequences of interest tend to be conserved. And alternatively, the difference Q-=QN-QC between summary charges at the N- and C-ends of alpha helices demonstrates greater variety if to compare with random sets (Figure 3b).
In general, our analysis has revealed the importance of charges at the N- and C-ends of a -helices of the basic region of bZIP domains. In our opinion, correlations between them may reflect the ability of a -helices to act as dipole. It might be very important for the interaction of those helices in dimers either with each other or with DNA. The detailed description of the results of correlation analysis is given in our previous work. To explain the above described correlations, we proposed a model of electrostatic interactions within the "DNA-basic region" complex (Afonnikov et al, 1997).
As for the matrix of partial correlation coefficients calculated using side chain volume quantities, most of significant elements were found nearby the main diagonal. It means that there are intensive steric contacts between the neighboring residues on the helix surface (including those located within the DNA-binding region of the helix).
Now, let us consider the difference between correlations as obtained by means of ordinary and partial coefficients. From the matrix diagrams (Figure 3) one can see that the difference is considerable for all the amino acid characteristics evaluated. And the more thorough analysis reveals the following interesting details. First, ordinary coefficient provides too many “false” correlations. In these cases correlation is considered as insignificant according to partial coefficient but treated as significant by ordinary one (for example see pair 222-224 for the correlation matrices of isoelectric point, Figure 3a). Second, there are pairs whose partial correlation coefficients are reliably non-zero, and the ordinary ones vanishes. These are “missed” correlations, as for pair 231-232 of the same matrices. Third, in some cases, significant ordinary and partial coefficients for the same pair of positions differ by their sign (“inversion”), e.g. pairs 225-228, 232-241 for isoelectric point matrices. It should be noted also that matrices of ordinary correlation coefficients for isoelectric point and hydrophobicity have no any ordered structure in comparison with those of the partial coefficients. Thus, the elements and the structure comparison of the matrices obtained by means of partial and ordinary coefficients indicates their considerable difference.
We suppose that superposition of a great number of real correlations in proteins might be responsible for thus revealed difference between matrices of the partial and ordinary correlation coefficients. It is known that proteins are characterized by the wide range of strongly interdependent variables (in particular, values of physical or chemical properties for amino acids in sequence positions). Ordinary correlation coefficient reflects a dependence between two positions as a result of superposition of direct interaction between them and a cumulative effect of their interactions with all the remained positions of a protein. That is why using of only ordinary correlation coefficient might result in inadequate detection of dependencies (“false”, “missed” and “inverted” correlations). At the same time, partial correlation coefficient gives a chance to eliminate these effects. However, even partial correlation coefficient reflects not only pairwise interactions between residues. There are other external factors not established in amino acid sequences directly (presence of ligands, multimeric protein complexes, physical and chemical conditions of the environment, etc.). Nevertheless, applying the partial correlation coefficient may serve as a useful tool to reveal such dependencies as well.
CONCLUSION
The suggested method estimates direct dependencies between values of chemical or physical properties of amino acids throughout position pairs of a multiply alignment. The approach allows to eliminate the influence of the remained sequence positions on the investigated pair. Therefore the above described technique might occur quite useful for detecting functional and structural features of the multiple aligned sequences. This could help to reveal connections between the primary sequences and structures of proteins with known spatial structure as well as of proteins encoded by newly discovered genes.
REFERENCES
Altshuh, D., Lesk, A.M., Bloomer, A.C., Klug, A. (1987) Correlation of co-ordinated amino acid substitution with function in viruses related to tobacco mosaic virus. J. Mol. Biol., 193, 693-707.
Anderson, T.W. (1958) An Introduction to Multivariate Statistical Analysis, Wiley, New York.
Afonnikov, D.A., Kondrakhin, Yu.V., Titov, I.I. (1997) Revealing of correlated positions of the DNA-binding region of the CREB and AP-1 transcription factor families. Molec. Biol.(Russian), in press.
Bairoch, A. and Boeckman, B. (1991) The SWISS-PROT protein sequence data bank. Nucleic Acids Res., 19, 2247-2249.
Chiu, D.K.Y., Kolodziejczak, T. (1991) Inferring consensus structure from nucleic acid sequences. Comput. Applic.Biosci., 7, 347-352.
Chothia, C. (1984) Principles that determine the structure of proteins. Ann. Rev. Biochem., 53, 537-572.
Clarke, N.D. (1995) Covariation of residues in the homeodomain sequence family. Protein Sci., 4, 2269-2278.
Eisenberg, D., Weiss, R.M., Terwilliger, T.C. (1984) The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA, 81, 140-144.
Ellenberger, T.E., Brandl, C.J., Struhl, K., Harrison, S.C. (1992) The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted a helices: crystal structure of the protein-DNA complex. Cell, 71, 1223-1237.
Gö bel, U., Sander, C., Schneider, R., Valencia, A. (1994) Correlated mutations and residue contacts in proteins. Proteins Struct Funct Genet., 18, 309-317.
Gutell, R.R., Power, A., Hertz, G.Z., Putz, E.J., Stormo, G.D. (1992) Identifying constraints on the higher-order structure of RNA: Continued development and application of comparative sequence analysis methods. Nucleic Acids Res., 20, 5785-5795.
Kolchanov, N.A., Titov, I.I., Vlassova, I.E., Vlassov, V.V. (1996) Chemical and computer probing of RNA structure. Progr. Nucl. Acid Res. Mol. Biol., 53, 131-196.
Korber, B.T.M., Farber, R.M., Wolpert, D.H., Lapedes, A.S. (1993) Covariation of mutations in the V3 loop of human immunodeficiency virus type 1 envelope protein: an information theoretic analysis. Proc. Natl. Acad. Sci. USA, 90, 7176-7180.
Landshultz, W.H., Johnson, P.F., McKnight, S.L. (1988) The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science, 240, 1759-1764.
Neher, E. (1994) How frequent are correlated changes in families of protein sequences? Proc. Natl. Acad. Sci. USA, 91, 98-102.
Seledtsov, I.A., Wolf, Yu.,I., Makarova, K. S. (1995) Multiple alignment of sequences based on the searching of statistically significant common regions. Mol. Biol.(Russian), 29, 1023-1039.
Shindyalov, I.N., Kolchanov, N.A., Sander, C. (1994) Can three-dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Eng., 7, 349-358.
Taylor, W.R., Hatrick, K. (1994) Compensating changes in protein multiple sequence alignment. Protein Eng., 7, 341-348.
White, A., Handler, P., Smith, E.L., Hill, R.L., Lehman, I.R. (1978) Principles of Biochemistry, vol. 1. McGraw-Hill, Inc.
FIGURE LEGENDS
Fig. 1. Amino acid sequence of the basic region of bZIP domain (a) and its three-dimensional structure in DNA-protein complex (b) for GCN4 transcription factor.
a). Low-variable positions not included in the analysis are underlined. Residues in contact with DNA are bold marked (according to Ellenberg et al, 1992).
b). Ribbon representation for structure of basic region bound to DNA. Basic region with its flanking residues for one of two monomers and DNA are shadowed.
Fig. 2. Diagrams of correlation coefficient matrices for isoelectric point (a), side chain volume (b) and hydrophobicity (c) quantities. Each diagram represents values of ordinary correlation coefficients (above the principal diagonal) and partial correlation coefficients (below the principal diagonal) for each analyzed pair of sequence positions. Significant positive (1>r>rc), significant negative (-1<r<-rc) and non-significant (|r|<rc) correlation coefficient values r are represented by white, black and dotted patters, respectively.
Fig. 3. Distributions of Q+ (a) and Q- (b) variance in 1000 samples of randomized sequences obtained via permutation test. Q+ and Q- values for real sample are shown by arrows.
Appendix 1.
List of CREB and AP-1 transcription factor families investigated. Identifier - ID and accession number - AC (in brackets) are given for each transcription factor.
AP1_CHICK (P18870), AP1_COTJA (P12981), AP1_DROME (P18289), AP1_HUMAN (P05412), AP1_MOUSE (P05627), AP1_RAT (P17325), AP1_SCHPO (Q01663), ATF1_HUMAN (P18846,P25168), ATF2_RAT (Q00969), ATF3_HUMAN (P18847), ATF4_HUMAN (P18848), ATF4_MOUSE (Q06507), ATFA_HUMAN (P17544), CAD1_YEAST (P24813), CREA_HUMAN (Q03060), CREA_MOUSE (P27698), CREB_BOVIN (P27925),CREB_HUMAN (P16220, P21934), CREB_MOUSE (Q01147), CREB_RAT (P15337), CREM_MOUSE (P27699), CREM_RAT (Q03061), CREP_HUMAN (P15336), FOSB_MOUSE (P13346), FOSX_MSVFR (P29176), FOS_AVINK (P23050), FOS_CHICK (P11939), FOS_HUMAN (P01100), FOS_MOUSE (P01101), FOS_MSVFB (P01102), FOS_RAT (P12841), FRA1_HUMAN (P15407), FRA1_RAT (P10158), FRA2_CHICK (P18625), FRA2_HUMAN (P15408), FRA2_MOUSE (P47930), FRA_DROME (P21525), GCN4_YEAST (P03069, P03068), HAC1_YEAST (P41546), JUNB_HUMAN (P17275), JUNB_MOUSE (P09450), JUNB_RAT (P24898), JUND_CHICK (P27921), JUND_HUMAN (P17535), JUND_MOUSE (P15066), LRF1_RAT (P29596), PDR4_YEAST (P19880, P22631), SKO1_YEAST (Q02100),TJUN_AVIS1 (P05411).


© 1997-99, IC&G SB RAS, Laboratory of Theoretical Genetics