GENEEXPRESS: A COMPUTER SYSTEM FOR DESCRIPTION, ANALYSIS, AND RECOGNITION OF REGULATORY SEQUENCES IN EUKARYOTIC GENOME
N.A. Kolchanov, M.P. Ponomarenko, A.E. Kel, Yu.V. Kondrakhin, A.S. Frolov, F.A. Kolpakov, T.N. Goriachkovskaia#, O.V. Kel, E.A. Ananko, E.V. Ignatieva, O.A. Podkolodnaya, V.N. Babenko, I.L. Stepanenko, A.G. Romashchenko, T.I. Merkulova, D.G. Vorobiev, S.V. Lavryushev, Yu.V. Ponomarenko, A.V. Kochetov, G.B. Kolesov,
Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090; Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia, 630090; email:kol@bionet.nsc.ru
V. V. Solovyev
The Sanger Centre Hinxton, Cambridge, CB10 1SA, UK email: solovyev@sanger.ac.uk
L. Milanesi
Istituto Di Tecnologie Biomediche Avanzate, Consiglio Nazionale Della Ricerche, Via Ampere 56, Milano, Italy; email: milanesi@icil64.cilea.it
N. L. Podkolodny
Institute of Computational Mathematics and Mathematical Geophysics, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia, 630090; email:pnl@omzg.sscc.ru
E. Wingender, T. Heinemeyer
Gesellschaft fur Biotechnologische Forschung mbH, Mascheroder Weg 1, D-38124 Braunschweig, Germany; email: ewi@gbf.de
Keywords: Gene networks, transcription, translation, regulation, site, recognition, activity, databases
.
Abstract
GeneExpress system has been designed to integrate description, analysis, and recognition of eukaryotic regulatory sequences. The system includes 5 basic units: (1) GeneNet contains an object-oriented database for accumulation of data on gene networks and signal transduction pathways and a Java-based viewer that allows an exploration and visualization of the GeneNet information; (2) Transcription Regulation combines the database on transcription regulatory regions of eukaryotic genes (TRRD) and TRRD Viewer; (3) Transcription Factor Binding Site Recognition contains a compilation of transcription factor binding sites (TFBSC) and programs for their analysis and recognition; (4) mRNA Translation is designed for analysis of structural and contextual features of mRNA 5’UTRs and prediction of their translation efficiency; and (5) ACTIVITY is the module for analysis and site activity prediction of a given nucleotide sequence. Integration of the databases in the GeneExpress is based on the Sequence Retrieval System (SRS) created in the European Bioinformatics Institute.
GeneExpress is available at /mgs/systems/geneexpress/.
Introduction
The eukaryotic gene expression is one of the most complex biological phenomena involving a number of molecular events. It may start with reception of a definite stimulus by the cell, which is then conveyed via the particular signal transduction pathway to initiate transcription of the relevant genes. Their pre-mRNAs are processed by 3’ cutting/polyadenylation, capping, splicing, and finally the corresponding proteins are translated from these mature mRNAs. This totality of molecular events forms the particular gene network that provides the cell response to the stimulus. The cellular and organismic homeostases as well as cell/tissue differentiation and development are maintained by their gene networks. That is the reason why molecular biologists investigating the gene expression should be able to access databases on all the stages of gene expression as well as the relevant programs for their analysis. Thus, investigation of the gene expression is an integrative problem of biology.
Currently, various experimental data on genomic regulatory sequences controlling the eukaryotic gene expression are being rapidly accumulated. The transcription regulatory regions have been sequenced for thousands of genes. A great number of transcription regulatory elements have been localized including transcription factor binding sites, enhancers, promoters, etc. (Kel’ A.E. et al., 1997; Peter R.C. et al., 1998; Wingender E. et al., 1996). A wide range of functional sites controlling other stages of gene expression (splicing, processing-polyadenylation, and translation) have been isolated and studied. A considerable volume of the experimental data on the activity of various types of functional sites controlling the gene expression has been generated (Kolchanov N.A. et al., 1998). The experimental data on the gene networks, the ensembles of coordinately functioning genes (Kolpakov F.A., et al., 1998), is growing. Computer analysis of the genomic regulatory sequences becomes even more important in case of functional interpretation of newly sequenced genomic fragments as well as for study of the molecular mechanisms of gene expression regulation. The current number of databases on various genomic regulatory regions is considerable. In addition to the general databases, such as EMBL and GenBank, a number of specialized databases on gene expression regulation are available: EPD (Peter R.C. et al., 1998), TRANSFAC (Wingender E. et al., 1996), TRRD (Kel’ A.E. et al., 1997), COMPEL (Kel O.V. et al., 1995b), EpoDB (Salas F. et al., 1998), etc. Many computer methods for recognition of regulatory genomic sequences (Waterman et al, 1984; Lawrence et al., 1993; Chen et al., 1995; Ulyanov & Stormo, 1995; Quandt K. et al., 1995; Fickett & Hatzigeorgiou, 1997 (review); Kel A.E. et al., 1995; Prestridge, 1995; Pedersen et al., 1996; Solovyev & Salamov, 1997; Salamov & Solovyev, 1997) have been developed. Thus, the challenging problem is to create a WWW-based environment capable of integrating the information coming from various databases on expression regulation and make this information accessible by software for investigation and prediction of regulatory sequences.
We started this integration from cross-linking the TRANSFAC, TRRD, and COMPEL databases through introduction of a common format table for all of them (Wingender et al., 1996). Appearance of SRS query system (Etzold and Argos, 1993) opened a new era of web-integration. It provides unification of queries to various databases concealing any specific details of their realization; unified representation of the queried information; flexible format of information representation (for example, FASTA, PIR, etc.); a possibility to include additional modules for graphic representation; a powerful reference and help systems for each of the databases; and a possibility of linkage with the other databases and computer systems.
Using SRS, we have developed GeneExpress, the SRS-based integrator for the databases and programs supporting investigation of the gene expression. The database GeneNet on molecular events forming gene networks was assigned its integrative core. To study transcription, this core was supplemented with the database TRRD on transcription regulatory regions and the compilation TFBSC of the sequence sets of transcription factor binding sites. The TRRD and TFBSC were linked to the system RgScan, recognizing the sites in DNA sequences. For translation, the database LeaderRNA on mRNA leaders was included and linked to the program predicting the High/Low translation levels from a given mRNA sequence. The gene expression is also quantitatively described by the system ACTIVITY compiling the functional site activity magnitudes and linked with the programs predicting the activities from site sequences. Thus, the GeneExpress system is designed to integrate description, analysis, and recognition of eukaryotic genomic sequences. The modular and hierarchical organization of regulatory genomic sequences and the network-organized regulation of gene expression were taken into consideration during the system development. GeneExpress, is WWW-available at /mgs/systems/geneexpress/.
GENE NETWORKS
The GeneNet database is designed for accumulation of formalized description of gene networks and signal transduction pathways. Using the object-oriented approach, the following components are included in the description of a gene network: entities (any material objects), relations between the entities, and processes connected with them (for example, viral infection, anemia, or erythrocyte differentiation). Four classes of entities are distinguished: (1) Cell (tissue, organ) entity, regarded as a definite compartment containing a certain set of entities of other classes; (2) Protein; (3) Gene; and (4) Substance (a nonprotein regulatory substance, for example, metabolite). Two classes of relations between the entities are described: (1) reaction of interaction between entities yielding a new entity or process; and (2) regulatory event as the effect of an entity on a certain reaction. Instances of Cell (tissue, organ), Gene, Protein, Substance, State, and Relation classes are described in the separate tables CELL, GENE, PROTEIN, STATE, and RELATION, respectively. The database is also supplemented with the SCHEME table. Thus, the database contains eight tables in the EMBL-like text format: (1) CELL (information on the cell types and lines, including also the description of tissues and organs); (2) GENE (genes and their regulatory features based on the information from the TRRD database); (3) PROTEIN (proteins and protein complexes); (4) SUBSTANCE (regulatory substances and metabolites); (5) PROCESS (physiological process and the organismic state during the gene network functioning); (6) RELATION (relations between the gene network components); and (7) SCHEME (description of the gene network graph).
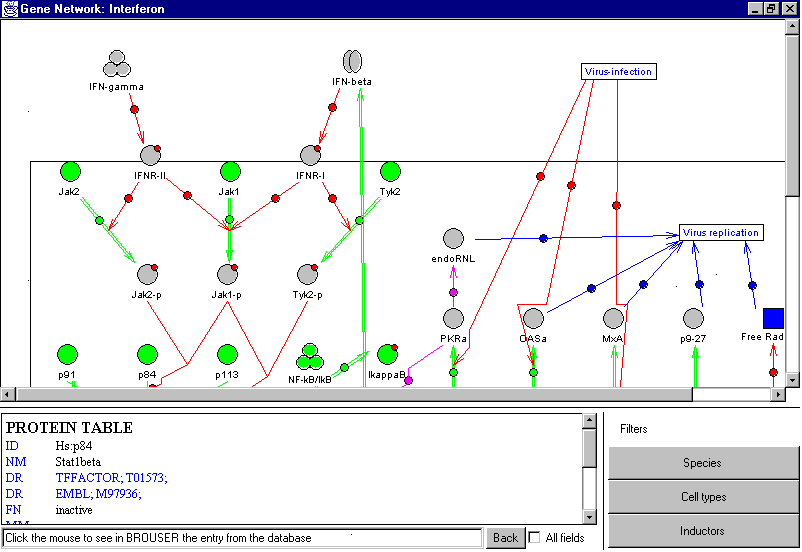
Figure 1. Example of automated construction of the diagram representing the gene network of the antiviral response at the cell level
The GeneNet database is also developed using the SRS. It supports the cross-references within the GeneNet database and with EMBL, SWISS-PROT, TRRD, TRANSFAC, and EPD databases. The current version of the GeneNet database contains the descriptions of gene networks of antiviral response (Ananko et al., 1997) and erythropoiesis (Podkolodnaya and Stepanenko, 1997).
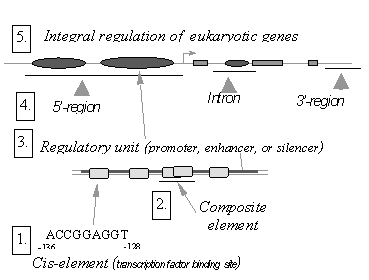
Figure 2. Structural and functional organization of eukaryotic genes.
The GeneNet includes automatic construction of a gene network diagram. The diagram is presented as a graph with the nodes corresponding to entities or states and the edges reflecting the relations between the gene network components. Information on the graph structure is taken from the SCHEME table. Each gene network component has its own image on the diagram, showing its features (Fig. 1). The GeneNet system takes into account that the gene network components can belong to different organs, tissues, cells, and cell compartments. The three following hierarchical levels are considered: (1) organism level, at which such entities as organs, tissues, cell types, and various substances affect other organs, tissues, and cells; (2) the single cell level, where four compartments are distinguished: the intercellular space, cell membrane, cytoplasm, and nucleus; and (3) the single gene level, where the description of transcription regulation employs the data from the TRRD database. Each level can be displayed in a separate window. The gene level is visualized via the TRRD Viewer described above.
The GeneNet Viewer is a Java applet. It includes the above-described generation of the gene network diagram and some tools for data navigation, on-line help, interactive cross-references within the GeneNet database, and references to other databases. All images on the diagram are interactive, i.e., if a user clicks the image, the textual description of the corresponding entry is displayed in the special text window under the diagram (Fig. 1). Double clicking the gene image starts the TRRD Viewer, and the regulatory map of the gene is visualized. The text window contains a formatted text with hypertext references of three types: (1) the reference explaining the type of information described in the field; (2) cross-references within the GeneNet database; and (3) references to other databases (EMBL, SWISS-PROT, TRRD, TRANSFAC, and EPD).
TRANSCRIPTION REGULATION
Transcription Regulatory Regions Database (TRRD)
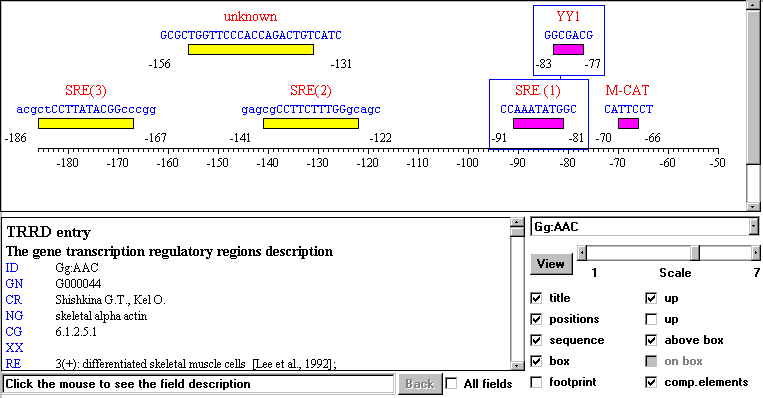
Figure 3. Example of visualization of gene regulatory map by TRRD Viewer. Boxes represent binding sites of transcription factors; the line connecting rectangles indicates a composite element.
The model of functional organization of eukaryotic gene regulatory regions (Kel’ A.E. et al., 1997; Kel O.V. et al., 1995a) was used as the basis for the TRRD database. It takes into account a great diversity of the elements controlling gene transcription, their modular organization, and the hierarchy of these elements, essential for their functioning. The TRRD format allows to describe the modular structure of transcription regulatory regions and the hierarchy of their constituent regulatory units. The hierarchy of the following elements has been implemented (Fig. 2): (1) Cis-elements provide the interaction of transcription factors with DNA (Wingender E., 1993); (2) Composite elements support the interactions between DNA sites and the protein factors or the protein-protein interactions, causing either synergistic or antagonistic regulatory effects (Kel O.V. et al., 1995b; Kel’ A.E. et al., 1997); (3) Promoters, enhancers, and silencers at this level of the hierarchy provide the transcription regulation under certain conditions; (4) Transcription regulatory regions are represented by continuous regions of genomic DNA containing the regulatory elements of the levels described above (Kel O.V. et al., 1995a; Kolchanov N.A., 1997) and located in the gene 3’- and 5’-flanking regions or introns; and (5) The system of integral regulation of gene transcription comprises all these regulatory elements (Kolchanov, N.A., 1997; Kel’, A.E. et al., 1997).
The TRRD database has three interconnected tables: TRRDGENES (description of genes), TRRDSITES (description of sites), and TRRDBIB (references).
TRRDGENES contains a general description of genes, peculiarities of their transcription regulation (dependence on the cell cycle stage, developmental stage, tissue-specificity, or effects of external factors), chromosomal location, description of regulatory units (promoters, enhancers, or silencers), composite elements, and free text comments. This table is linked to the TRANSFAC_GENE (Wingender E. et al., 1996), COMPEL (Kel O.V., 1995b), and GeneNet (Kolpakov F.A. et al., 1998) databases.
TRRDSITES accumulates information on transcription factor binding sites: nucleotide sequences and their location within the gene, the list of the relevant cell lines and codes of experiments, and free text comments. This table is linked to the following computer systems: ACTIVITY for predicting site activity (Kolchanov N.A. et al., 1998) and programs for site recognition (Kondrakhin Yu.V. et al., 1998). The table also contains the references to TRANSFAC_SITE, TRANSFAC_FACTOR (Wingender E. et al., 1996), and EMBL databases.
TRRDBIB is linked with the site and gene description tables and contains the complete references to the original articles and to MEDLINE references.
The current version, TRRD 3.5, comprises the description of 427 genes, 607 regulatory units (promoters, enhancers, and silencers), and 2147 transcription factor binding sites. Over 1500 scientific publications have been processed to obtain these data. The TRRDGENES database includes information on human (185 entries), mouse (126), rat (69), chicken (29), and other genes. Most of the tissue-specific genes are expressed in liver (120), blood (67), or muscle cells (37). The major part of the genes compiled in TRRD are either interferon-induced (62) or glucocorticoid-regulated genes (30), or belong to lipid metabolism (41), erythroid differentiation (37), or cell cycle regulation (23) functional systems. TRRD is installed under the SRS to provide easy information retrieval and integration with other databases and computer systems for information processing.
TRRD Viewer. The Java applet, TRRD-Viewer, allows to visualize the data on location of transcription factor binding sites in a map form (Fig. 3) and overlook their textual description. While working with this applet, the user selects a gene identifier from the list, and the textual description of the gene (from TRRDGENES), its sites (from TRRDSITES), and the relevant references (from TRRDBIB) appears in the text window. Transcription factor binding sites and composite elements are presented graphically. If the user clicks the site image, the description from the TRRDSITES table is displayed in the text window. Clicking the field title provides comments on the information described in the field. Several options allow a number of different site representations.
*********************************************
* Prediction of potential binding site
* of transcription factors in a given
* DNA sequence on the basis of
* recognition groups.
*******************************************
The name of sequence: XLACTA2 standard;
The number of all predicted binding sites = 14
Name Position Site
APF 5 agtaac
NFIII 52 tcattt
GT-2B 81 cagctg
MLTF 107 gtcact
AP-1 108 tcactca
SRF 221 ccatgtaagg
GATA-1 262 cttatca
SRF 272 ccaaatatgg
NFIII 288 aaatga
AR 301 tcttct
AR 356 agagca
NFIII 361 aaatga
Pit-1 378 atgaata
TFIID 385 tataaa
Figure 4. Prediction of transcription factor binding sites in the promoter region of Xenopus laevis alpha2 gene (sarcomeric actin) by SiteScan program.
TRANSCRIPTION FACTOR BINDING SITE RECOGNITION
Table 1. A set of realizations for AP-1 binding site
N |
Weight $) |
Realization |
0 |
26 |
tgactca |
1 |
10 |
tgactAa |
2 |
5 |
tgaAtca |
3 |
4 |
tgacGca |
4 |
2 |
tAactca |
5 |
2 |
tgacAca |
6 |
2 |
tgactGa |
7 |
1 |
tCactca |
8 |
1 |
TgGctca |
9 |
1 |
TgactcG |
10 |
1 |
TgactcC |
$) Realization weight is the number of binding sites from U0 containing a given realization.
This module includes two blocks: the database on transcription factor binding site compilations (TFBSC) and programs for site analysis and recognition SiteGroup and SiteScan (Kondrakhin Yu.V. et al., 1998). The training samples from TFBSC, containing experimentally determined sequences of a particular site have been used in developing the recognition methods. The data include samples of 41 transcription factor binding sites (from 6 to 199 sequences for each factor; 1496 sequences totally) in EMBL-like format.
Table 2. Examples of the accuracy of binding site recognition
by SiteScan program
No |
Binding |
Errors*) for RGScan method |
Errors for matrix method |
||
site |
a 1 |
a 2 |
a 1 |
a 2 |
|
1 |
AP1 |
0.188 |
0.004303 |
0.156 |
0.007421 |
2 |
AP2 |
0.125 |
0.000872 |
0.063 |
0.032477 |
3 |
ATF/CREB |
0.147 |
0.000207 |
0.118 |
0.000964 |
4 |
C/EBP |
0.060 |
0.023392 |
0.096 |
0.021284 |
5 |
COUP/RAR |
0.025 |
0.003936 |
0.050 |
0.057874 |
6 |
ETF |
0.000 |
0.002229 |
0.333 |
<10-5 |
7 |
GATA |
0.127 |
0.000491 |
0.072 |
0.015774 |
8 |
GR |
0.118 |
0.003092 |
0.039 |
0.033286 |
9 |
NF-1 |
0.038 |
0.000620 |
0.099 |
0.013694 |
10 |
NF-kB |
0.138 |
<10-5 |
0.069 |
0.002014 |
11 |
NF-Y |
0.045 |
0.001466 |
0.409 |
0.000258 |
12 |
OCT |
0.163 |
0.000505 |
0.571 |
<10-5 |
13 |
Pit-1 |
0.176 |
0.000522 |
0.059 |
0.009959 |
14 |
Sp1 |
0.029 |
0.008347 |
0.194 |
0.001999 |
*)
a 1, false negatives; a 2; false positivesA simple recognition method is based on the representation of transcription factor binding sequences as a set of site realizations R={R0, R1, ..., Rk-1}. In other words, the set of realizations in the form of oligonucleotides t bases long coded in the IUPAC-IUB 15 single-letter based codes is created by our algorithm for any particular site. This approach avoids the averaging of the nucleotide composition, as occur while describing the functional site by the weight matrix or consensus. The program SiteGroup uses a set U0={u1,...,um} of experimentally determined binding site sequences extracted from the TFBSC as the initial information to create the set of realizations R. It is determined by two parameters: (1) the length of the oligonucleotide t and (2) the maximally allowable difference (distance) t(miss) between these oligonucleotides. The main realization is the oligonucleotide R0 of length t having the highest frequency in the sample U0. Then, all sequences ui containing R0 are removed from the sample U0, producing the sample U1 and so on. Analysis of the sample Ur, during which the rth realization Rr is searched for, is performed at the rth step. In this iterative process, all oligonucleotide words of length t are considered. The distance t from the word R0 is estimated for each word. The word Rr that has the least distance from the R0 word is selected. If a number of words have the same minimal distance, the word with the maximal frequency in the set Ur is selected. The iteration is stopped when the set Ur either becomes empty or contains only the words that differ from R0 by the value exceeding t(miss). Each set of realizations created by the method described may be characterized by parameter fij: the proportion of the sequences from the initial set U0 represented in this set of realizations (covering of the set U0). The set of realizations providing maximization of the functional is searched for by exhaustion of the pairs (t(miss),t )=(i,j).
Expert Weights (0-10 are valid; 5 employed automatically)
Figure 5. List of 5’UTR mRNA characteristics important for predicting the level of gene expression. Parameters 8-12 were determined for the (-35;-1) 5’UTR fragment.
qij=fij* [(fij-fij-1)+(fij-fij+1)]. (1)
Examples of the first and second type errors in recognition of several transcription factor binding sites are listed in Table 2. For assessing the second type errors, the compilation of eukaryotic non-first exons was used. Small first and second type errors were observed for the SiteScan recognition. It is especially important for analysis of transcription factor binding sites in long genomic sequences.
COMPUTER SYSTEM FOR PREDICTING mRNA TRANSLATION ACTIVITY
This part of the GeneExpress system is designed for prediction of mRNA translation efficiency basing on analysis of structural and contextual features of 5’ untranslated regions (5’UTRs). It has a program (Leader) for mRNA translation rate prediction and the database containing 5’UTR sequences (Leader_Sq) and some information on the effect of several 5’UTR features on mRNA translation efficiency.
Eukaryotic mRNAs differ considerably in their translation efficiency. This has been attributed to different efficiency of translation initiation. The contextual and structural features of 5’UTRs have strong effect on translation initiation. To reveal these characteristics, we have compared the mRNA sequences of several house-keeping gene groups, highly expressed in eukaryotic cells, and some groups of regulatory genes, whose expression is low and under stringent control. The group of highly expressed mRNAs consists of mRNAs of highly abundant proteins such as actins, tubulins, ribosomal proteins, histones, hsp70, etc.
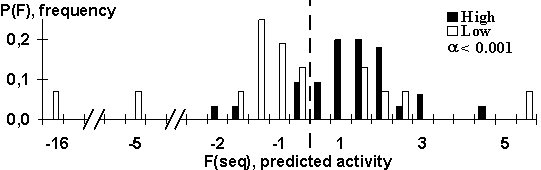
Figure 6. The control results were obtained using a set of independent data. Broken line is the selected threshold to separate low and high expressed mRNA.
Low expression mRNAs include mRNAs of transcription factors, protein kinases, growth factors, protooncogenes, etc. We have found several features that are different for these two groups (Fig. 5): 5’UTR length, nucleotide composition, context of start AUG codon, and presence of AUGs within 5’UTRs (Ischenko I.V. et al., 1996; Kochetov A.V. et al., 1998). These 5’UTR features affect the 40S ribosomal subunit movement along the leader and, therefore, the efficiency of the translation initiation.
The difference in these features was used for discrimination between high and low expressed genes The program calculates the 5’UTR features and evaluates the translation activity of mRNA. We create a simple discrimination function based on Penrose distance. The discrimination between the control samples of the high and low expressed mRNAs of dicot plants showed that 84% of the high and 76% of the low expressed mRNAs were classified correctly (Fig. 6).
SEQUENCE-BASED PREDICTION OF FUNCTIONAL SITE ACTIVITY
Initial postulates. It is suggested that the site activity F is determined by context-dependent properties of its nucleotide sequence S: statistical, physical, and conformational (M.P. Ponomarenko et al., 1997a, N.A. Kolchanov et al., 1998). These properties are of two types (A.E. Kel et al., 1993): (1) obligatory, which are invariant for all sequences Sn of the site and determine its basal activity; and (2) facultative, which are individual in terms of their “number, size, and location” for each sequence of the site and modulate the site activity with respect to the basal level. Hence, within the framework of the linear-additive approximation, the activity of the site with sequence S may be described by the following equation:
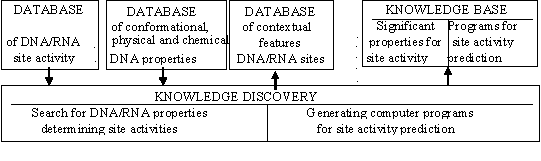
Figure 7. Principal scheme of the ACTIVITY computer system.
![]() ; (2)
; (2)
where F0(S) is the basal activity level determined by the occurrence of the obligatory properties of this site in the sequence Sn; {Xk}k=1,K are the facultative properties; and Fk is the contribution of the facultative property Xk to the site activity F. The principal scheme of the ACTIVITY system is shown in Fig. 7.
MI P0000001
MN Conformational
MD B-DNA
ML dinucleotide step
HN SCI00001
RN RF000012
RN RF000017
PN Twist
PM Calculated
PV TwistCalc
PU Degree
DINUCLEOTIDE
AA 38.90
AT 33.81
AG 32.15
AC 31.12
TA 33.28
TT 38.90
TG 41.41
TC 41.31
GA 41.31
GT 31.12
GG 34.96
GC 38.50
CA 41.41
CT 32.15
CG 32.91
CC 34.96
Figure 8. Description of conformational property “Helical twist angle of B-DNA” in the ACTIVITY system database.
Database on functional site activities compiles the available data on functional sites with the experimentally measured activities: over 240 site samples of different types, such as promoters and binding sites for E. coli regulatory proteins, TATA boxes and binding sites for eukaryotic transcription factors, translation starts, splicing and 3’-processing sites, etc. Site activity characteristics include the association/dissociation rates of DNA-protein complexes, their lifetimes, equilibrium constants, transcription and translation efficiencies, etc.
Database on conformational and physical/chemical DNA properties compiles the information on context-dependent properties which may play a significant role in DNA-protein interactions. The format of this database is illustrated in Fig. 8. The database currently contains over 40 conformational parameters, determined either computationally or by X-ray analysis. Over 10 physical DNA properties -- melting temperature, persistent length, bending-rigidity, entropy, etc. -- are also included. The system for knowledge discovery on site activity contains two blocks. The first is responsible for revealing any site properties significant for predicting the site activity and the second provides generation of C-code programs to predict the activity of a given sequence.
One example of the context characteristics of the sequence S is the positional weighted concentration Xzmw(S) of mono-, di-, tri-, and tetranucleotides (Ponomarenko et al., 1997b):
![]() , (3)
, (3)
where d Z is the "1" or "0" indicator function depending on the match or mismatch between the sequence S and the oligonucleotide Z; w(i) is the function of position effect determined according to the rule: “the more important is the position i for the site activity, the higher its assigned weight w(i)”. The activity prediction employs 180 various weight functions w(i). They are stored in the database for contextual characteristics of the ACTIVITY system (Fig. 9). Three examples of weight functions w(i) are shown in Fig. 10. The nucleotide composition of any oligonucleotide is presented in the 15 single-letter based code.
We also use the mean values of site S properties within the region [a;b] as site characteristics:
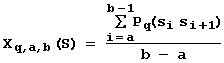 . (4)
. (4)
Search for statistically significant characteristics is implemented as analysis of conformational and physical characteristics for all oligonucleotides with lengths from 1 to M, checking each of the 180 position effect functions w(i). The total number of combinations <Z, m, w> is about 107 for m=4. Similarly, for every conformational or physical property q, the analysis of all possible locations of the region [a, b] within the site sequence is performed. Xqab(Sn) is calculated for a fixed combination <q, a, b> for each sequence of the site. The total number of the combinations <q, a, b> is 105.
The significance of a property for site activity prediction is estimated (Ponomarenko et al., 1997b) within the frames of the Utility Theory for Decision Making (P.C. Fishburn et al., 1970). Let’s calculate the fixed property Xzmw(Sn) for each sequence Sn with the known activity Fn. If the resulting pairs {Xzmw(Sn), Fn} meet all the necessary conditions of the linear regression applicability, then the activity F is predictable from an arbitrary sequence S via the feature Xzmw. To test these conditions of linear regression applicability, a simple regression is optimized for the pair {Xzmw(Sn), Fn}:
![]() ;(5)
;(5)
where f0 and f1 are the regression optimized coefficients.
To ensure the reliability of the regression between Xzmw(Sn) and Fn, 22 conditions of regression analysis are tested (the presence of linear, sign, and rank correlations between the predicted Fzmw(Sn) and the experimental Fn activities; the equality of Xzmw(Sn) and Fn distributions, etc.).The significance level a rt at which the rth condition is met is estimated. Then, the partial utility of the feature XZmw in predicting the activity F is calculated as follows:
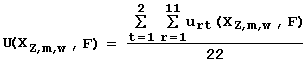 . (5)
. (5)
urt in the Utility Theory for Decision Making is determined as:
urt(XZmw,F)=![]() (6)
(6)
MI K0000039
CF SEQUENCE-DEPENDENT CONFORMATIONAL FEATURE
CT PROPERTY AVERAGED FOR REGION [A;B]
DP P0000001
PV Twist
AB 10 18
UT 0.234
LC -0.859
FG http://wwwmgs.bionet.nsc.ru/.../USFTWIST.htm
C-CODE
/*USF/DNA-binding increases with Twist decrease*/
double TwistCalc_for_SynthUSFbind (char *s){
double X; char *seq; int i,k, SiteLength=9;
double DinucPar[16]={ 38.90, ........, 34.96 };
seq=&s[0];
if(strlen(seq) < SiteLength+1)return(-1001.);
for (i=0, X=0.;i < SiteLength-1;i++) {
switch (seq[i ]) { case 'A': k= 0; break;
..........................................
switch (seq[i+1]) { case 'A': k+=0; break;
............................................
if (k > 15) return(-1004.); X+=DinucPar[k]; }
return (X/(double)(SiteLength-1));}
Figure 9. Description of a conformational property Helical twist for USF-binding site in the ACTIVITY.
Only the properties with U(XZmw,F)>0 are selected for the activity prediction and used to choose a limited set of linearly independent properties
a)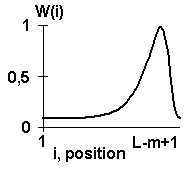
b)
c)
Figure 10. Examples of weight functions w(i).
Knowledge base on functional site activity. All selected characteristics are stored in the knowledge base of the ACTIVITY system. The format of the conformational property description is illustrated in Fig. 9. Using these properties, we generate the program to predict the site activity from its nucleotide sequence through optimization of equation (2). C-code programs for such prediction are also stored in the knowledge base (Ponomarenko et al., 1997b).
Analysis of several site samples has demonstrated applicability of this simple approach. For all these samples, the significant features have been identified and the linear-additive approximation for predicting the site activity has been derived. For example, the weighted concentration of the tetranucleotide VUKK downstream of the SV40 pre-mRNA cleavage point was found to be responsible for the 3’-processing efficiency (Fig. 11a). The USF/DNA factor affinity correlates very well with the helical twist of B-DNA (Fig. 11b). The major groove width determines the Cro repressor/DNA affinity (Fig. 11c). The identified characteristics provide a first approximation in predicting the value of the specific activity of functional sites. These properties were used to generate the method for predicting the transcription activity of mouse a A-crystalline gene by analyzing its promoter sequence; the prediction shows a good agreement with the experimental data (Fig. 11d).
a)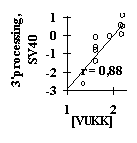 b)
b)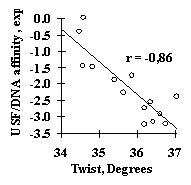 c)
c)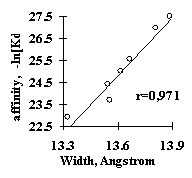 d)
d)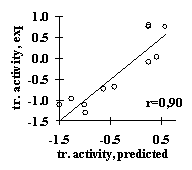
Figure 11. (a) Dependence of the mature DNA yield in the 3’-processing of SV40 virus pre-mRNA on the weighted concentration of VUKK tetranucleotide downstream of the mRNA cleavage point; (b) the dependence of the USF/DNA affinity on the helical twist angle of B-DNA; (c) the dependence of the Cro repressor/DNA affinity on the major groove width of B-DNA; (d) correlation of the experimental and calculated transcription activity for TATA/PE1B-containing promoter region of mouse a A-crystalline gene.
Reliability of the functional site activity values predicted by ACTIVITY from their sequences was studied by the authors earlier (Ponomarenko et al., 1997b) as well as ACTIVITY was compared with weight matrices (Stormo et al., 1986) and neural networks (Jonson et al., 1993).
CONCLUSION
The first step in interpreting a human genome sequence involves finding and annotation of the all genes it contains. The second step consists in characterizing the biological function of the individual genes, the way they are controlled, and their possible involvement in human disease. Significant success has been made in predicting and annotating protein coding regions (exons), although the exons account for only a few percents of the genomic sequence. A considerable part of the genome is occupied by regulatory sequences, which specify the tissue, developmental stage, or biochemical context of gene expression. Recognition, interpretation, and annotation of genome regulatory sequences should be one of the major tasks in the future progress of Human Genome Project. We designed the GeneExpress computer system as a first attempt to integrate the variety of information on genomic regulatory sequences and to use this information in developing and running software for their analysis and recognition. GeneExpress integrates TRRD and GeneNet databases and provides references to external databases, such as TRANSFAC, COMPEL, and EMBL using the SRS query system.
It is essential that the GeneExpress can and have to progress and expand continuously to update and integrate new resources for investigating other molecular events of the gene expression, such as splicing, DNA/protein interactions, etc. In the nearest future, a number of new basic modules will be added to the system including programs for recognition of eukaryotic promoters (Solovyev & Salamov, 1997) and composite regulatory elements (Kel et al., 1995). A great number of software and information resources on various aspects of gene expression regulation, developed by the bioinformatics community, are currently existing. However, representation diversity of the data and the results of the data processing hinders the access to these resources. This diversity is and will be the natural trait of bioinformatics, inherent for its development. Hence, the problem is not to develop uniform data formats but to succeed in integration of the already available software and information resources in the formats developed by their authors to make these resources maximally convenient for experimenters. The advent of the SRS (Etzold and Argos, 1993) opens a way to solve this problem. In addition, the users should be provided with the possibility to arrange complex scenarios of step-by-step running programs in the course of data analysis using the integrated WWW resources. This may be realized, for example, by creating a virtual knowledge base, so that the user can accumulate the results of analysis, visualize them, and compare to both one another and the information available in the integrated databases. In the project AUTOGENE (Ptitsyn et al, 1996), the authors have already tried to integrate the analyzing programs into flexible scenarios with input/output transfer of the results using the virtual knowledge base and demonstrated that the approach is promising.
Acknowledgments
This work was supported partially by grants of Russian National Human Genome Project, Russian Ministry of Science and Technical Politics, Siberian Department of Russian Academy of Sciences, and Russian Foundation for Basic Research (97-04-49740-a and 96-04-50006).
References: