Published in:
Computer Science and Biology. Proc. of German Conference on Bioinformatics, GSB’96 (Eds: Hofestadt et al), Liebigstr, Germany, 1996COMPUTER ANALYSIS AND RECOGNITION OF THE TRANSCRIPTION REGULATORY ELEMENTS IN EUKARYOTIC GENOMES
Kondrakhin Yu.V.1, Kolpakov F.A.1, Kel A.E., Milanesi L.2, Kolchanov N.A.1
1 Institute of Cytology and Genetics, Russian Academy of Sciences,
Siberian Department, Novosibirsk, Russia
2
Instituto di Tecnolologie Biomediche Avanzate,Consigilo Nazionale delle Richere, Milan, Italy
1
Tel: (3832)351263 Fax: (3832)353335 E.mail: kol@.bionet.nsk.su
Keywords: promoter recognition, transcription factor binding sites, composite element.
A feature of transcription regulatory regions of eukaryotic genes is a clear-cut hierarchy and modular structure (Kel, at al., 1995a). Binding sites for individual transcription factors are at the bottom. Composite elements are at the intermediate level; these are composed of closely located (adjacent or even overlapping) transcription factor binding sites that function as one by virtue of intensive protein-protein interaction between the transcription factors. At the highest level are promoters and enhancers. On the background of the projects for total sequence of the human, mouse, Drosophila and other eukaryotic genome, computer analysis and identification of transcription regulatory units is an extremely important problem. We report results of computer analysis and identification of transcription factor binding sites, composite elements, promoters.
Recognition of transcription factor binding sites by consensus. We developed FUNSITE-SIGNAL, a software program (Kel at al, 1995b), for identification of potential binding sites by using consensus. In searching for sites totally matching consensus, the second type error is as a rule low enough. By contrast, the first type is considerable and lies between 25% and 100% (Table 1). This is, however, lower if mismatches are allowed. As the number of mismatches is growing higher, the lower the first type error, the higher the second type error (Table 2).
Table 1. Typical first and second type errors, a 1, a 2, in identification of cis-elements by consensus.
Cis-element |
Consensus |
a 1 (%) |
a 2 (%) |
| AP1 | tgastma |
44 |
0.0221 |
| AP2 | cccmnsss |
52 |
0.4934 |
| AR | agaacannntgttct |
100 |
<0.001 |
| GATA-1 | wgatar |
33 |
0.1015 |
| NF-1 | yggmnnnnngccaa |
23 |
0.0040 |
| Oct-1 | atgcaaat |
75 |
0.0023 |
| Sp1 | Krggckrrk |
56 |
0.0645 |
Table 2. Dependence of first and second type errors, a 1, a 2, în the number of allowable mismatches t admiss in consensi
AR |
Sp1 |
NF-1 |
||||
t admiss |
a 1 (%) |
a 2 (%) |
a 1 (%) |
a 2 (%) |
a 1 (%) |
a 2 (%) |
0 |
100 |
0,000 |
55,6 |
0,065 |
23,1 |
0,004 |
1 |
100 |
0,000 |
11,1 |
1,198 |
11,5 |
0,076 |
2 |
95,0 |
0,009 |
0,0 |
7,118 |
5,8 |
0,847 |
3 |
67,5 |
0,077 |
3,8 |
4,976 |
||
4 |
48,8 |
0,396 |
3,8 |
19,204 |
||
5 |
25,0 |
1,592 |
1,9 |
47,100 |
||
6 |
12,5 |
5,680 |
||||
We developed a method which allows somewhat better accuracy. Under this method, any site is represented by a set of real oligonucleotides R={RÇ ,R1,...,Rk-1}, where any Ri is an oligonucleotide word t symbols in length. Any Ri, cannot have more than P mismatches. R was generated on the basis of the sample UÇ ={u1,...,um}, where ui is the binding site for the same transcription factor; the binding sites were from the Transcription Factor Database (TFD). The algorithm of sampling optimizes the parameters r and P so that R covers UÇ most efficiently. A typical example of the set R generated by this algorithm for site AP1 is presented in Table 3.
Table 3. Cis-element AP1 as a set of real oligonucleotides
Oligonucleotide |
Oligonucleotide No. |
Occurs in the set, times |
tgactca |
0 |
26 |
tgactAa |
1 |
10 |
tgaAtca |
2 |
5 |
tgacGca |
3 |
4 |
tAactca |
4 |
2 |
tgacAca |
5 |
2 |
tgactGa |
6 |
2 |
tCactca |
7 |
1 |
tgGctca |
8 |
1 |
tgactcG |
9 |
1 |
tgactcC |
10 |
1 |
A new method for site recognition, implemented in the software program
FUNSITE-SIG-REAL, a DNA fragment of length![]() is regarded as a site
provided it has a match in R. For most of sites, if the second type error is the same or
lower, the first type error is considerably lower following this method than the consensus
method (Table 4).
is regarded as a site
provided it has a match in R. For most of sites, if the second type error is the same or
lower, the first type error is considerably lower following this method than the consensus
method (Table 4).
Table 4. Comparison of errors of recognition by consensus and the set of real oligonucleotides
cis-elements |
Recognition by consensus |
Recognition by the set of real oligonucleotidesĺ |
||
a 1 (%) |
a 2 (%) |
a 1 (%) |
a 2 (%) |
|
AP-1 |
44 |
0,02 |
19 |
0,13 |
AP-2 |
52 |
0,49 |
44 |
0,07 |
GATA1 |
33 |
0,10 |
27 |
0,03 |
Oct-1 |
75 |
<0,01 |
14 |
0,03 |
Revealing composite elements. We suggest a new method for identification of composite elements, which includes: 1) identification of potential binding sites in a promoter sample; 2) identification of pairs of sites (A,B,w) no farther than w=50 bp apart; 3) statistical analysis of frequencies for all sites and pairs. (Ŕ,Â,w) is regarded as a composite element provided its observed frequency exceeds significantly (a <0.05) the expected frequency. With the aid of this approach, we have revealed more than 70 new composite elements. The {GATA-1, NF-kB} composite element occurring in a variety of promoters is presented in Fig.1.
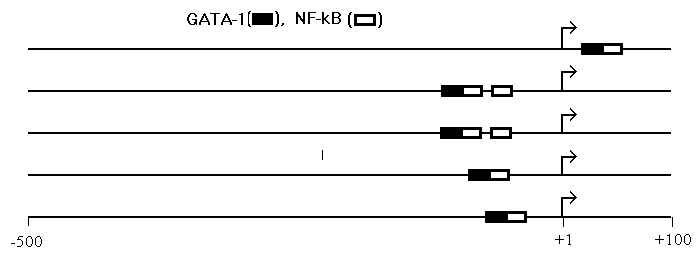
Fig.1. A composite element, {GATA-1, NF-kB}, identified in 5 promoter regions. Arrows indicate transcription start sites.
A promoter recognition method. The method depends upon the
concentration of binding sites and the unevenness of their distribution along the
promoters. To take account of the two parameters, each promoter region of length L= 600 bp
was divided into 80 fragments nearly the same in length. By analysis of the promoter
sample, we generated the matrix T, whose element ![]() was the observed
number of potential sites of type i (i=1,...,130) in the j-th region (j=1,...,80).
Fragment S of length L of the nucleotide sequence under study was regarded as a promoter
if m >m*, where m* is a fixed threshold value and
was the observed
number of potential sites of type i (i=1,...,130) in the j-th region (j=1,...,80).
Fragment S of length L of the nucleotide sequence under study was regarded as a promoter
if m >m*, where m* is a fixed threshold value and
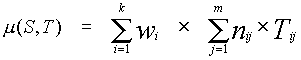 .
.
Here k =130 is the number of sites under consideration, nij is the total number of the I-th cis-element in the j-th region of fragment S. The weight coefficient wi of the i-th cis-element is defined as wi = -ln(P(i)), where P(i) is the probability of the I-th cis-element occurring in a random nucleotide sequence. In fact, the measure m is characteristic of fragment S containing eukaryotic promoters. By calculating m for each position of the sliding window 600 bp in length, it is possible to plot a curve for the transcription regulatory potential of any nucleotide sequence of eukaryotic genome. Fig.2 exemplifies the potential for a 12847 bp fragment of the human a-globine gene cluster. As one can see, the transcription regulatory potential peaks at the transcription start sites of two globine genes, while there is no such maximum before the pseudogene.
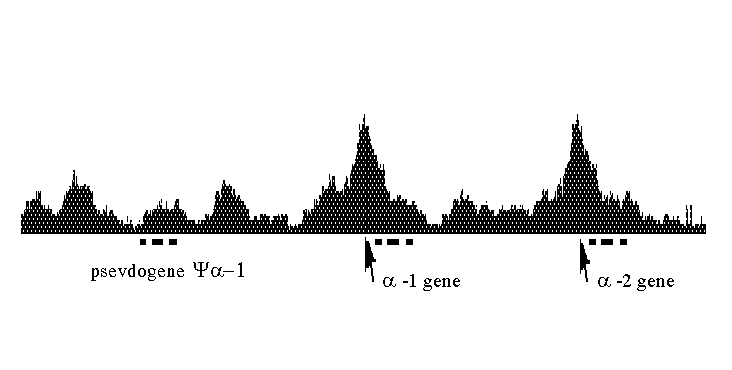
Fig. 2. Transcription regulatory potential for a fragment of the cluster of human a -globine genes (EMBL entry name - HSHBA4).
Some additional accuracy was gained by dividing the initial promoter
sample into 8 subsamples. Each contains promoters with similar distributions of binding
sites along the promoters. Fragment S was regarded as a type i (i=1,...,8) promoter given
that ![]() >
>![]() . Here the first type error of
promoter recognition was 35%, the second type error was just 0.8% (Kondrakhin et al,
1995).
. Here the first type error of
promoter recognition was 35%, the second type error was just 0.8% (Kondrakhin et al,
1995).
Acknowledgments
This work was supported in part by grant 94-04-13241 and 95-04-12757 from the Russian Fond of Fundamental Research. The authors are also thankful to V.Filonenko for translating this report from Russian into English.
References
Kel O.V., Romachenko A.G., Kel A.E., Naumochkin A.N., and Kolchanov N.A. (1995a). Data representation in the TRRD - a database of transcription regulatory regions of the eukaryotic genomes.//Proceedings of the 28th Annual Hawaii International Conference on System Scienses [HICSS]. Biotechnology Computing, IEE Computer Society Press, Los Alamitos, California, vol.5 pp. 42 -51.
Kel A.E., Kondrakhin Y.V., Kolpakov Ph.A., Kel O.V., Romashenko A.G., Wingender E., Milanesi L., Kolchanov N.A. (1995b). Computer tool FUNSITE for analysis of eukaryotic regulatory genjmic Sequences.//Proceedings third International conference on intelligent systems for molecular biology, California, US, pp. 197-205.
Kondrakhin Y.V., Kel A.E., Kolchanov N.A., Romashchenko A.G., Milanesi L. (1995). Eukaryotic promoter recognition by binding sites for transcription factors.//CABIOS, vol.11, pp. 477-488.