|
ANALYSIS OF INTEGRALCHARACTERISTICS[Tutorial page]
|
|

|
| As the integral
protein characteristics, the summarised charge Q+ of a group of residues in the region of
interaction of alpha-helices H1 and H2 of homeodomain (in terms of isoelectric point
value) is considered. The package CRASP estimates: - the impact of co-ordinated
substitutions into the constancy of Q+ value Denotations: |
|
| Open new browser window integral
characteristics analysis CRASP page. This will be the CRASP package
working window. Use it in parallel with this tutorial window. Open new browser window Result example page to retreive the data from test
analysis. The description of analysis, methods and algorithms could be obtained here. |
|
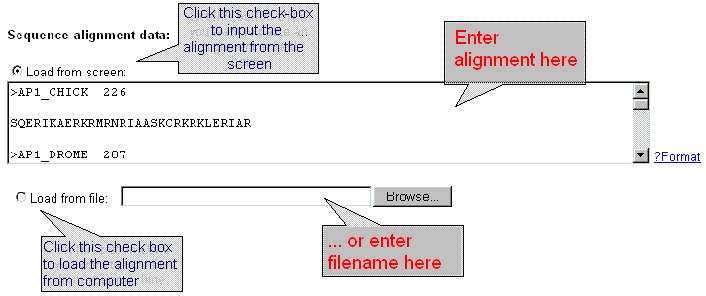
| Enter the sequence alignment data in FASTA format into the 'Sequence data' field or download the sequence data from file by using dialog window 'Load from file'. Click the button 'ON' and input the file name into the text field. |
 |
Your
actions with the working window:
|
|
| Select calculation parameters in drop-down menus and editable text fields: |
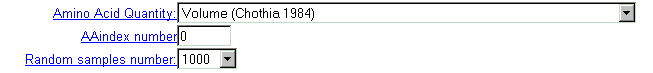 |
| Select physico-chemical residue's characteristics from the 'AminoAcid
quantity' menu, which contains 36 properties.
Important note: this option is valid if 'AAindex number' field contains zero value. |
 |
| You may select one of more than 400 characteristics from AAIndex database (see details). Type the database entry number. |
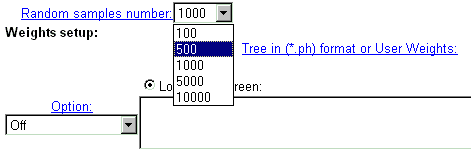
| In CRASP, Monte Carlo technique is applied to estimate the statistical significance of the constancy of the chosen physico-chemical parameters. For this purpose, a large number of random samples with independent amino acid substitutions in alignment positions is generated and folowed by subsequent evaluation of statistical parameters of dispersions of integral characteristics in these samples. The number of samples generated is ordered by menu 'Random samples number'. For the characteristic including many positions and for huge sequence samples, the calculations are time-consuming, so we recommend to use small values for this parameter. |
 |
| Your actions with the
working window: Set the following calculation parameters:
|
|
| Sometimes it is necessary to apply data weighting. CRASP package realises several standard schemes of data weighting. Also, a user may enter other weights for all sequences. Data weighting is set in the fields below. |
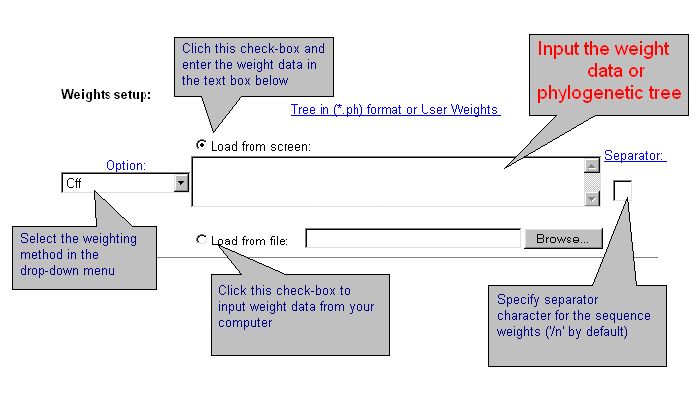 |
| Current version of CRASP applies several weighting methods. Select one of them or the option without weighting 'Off'. By choosing 'User defined' weights, enter weight values for each sequence that should be divided by symbol-separator (by default - ;). You may introduce your own symbol-separator, specified in the text-box 'Separator'. By using the weights by Altschul et al. and Felsenstein, enter the phylogenetic tree in *.ph format or load it from file. Details are here. |
 |
Your actions with the
working window:
|
|
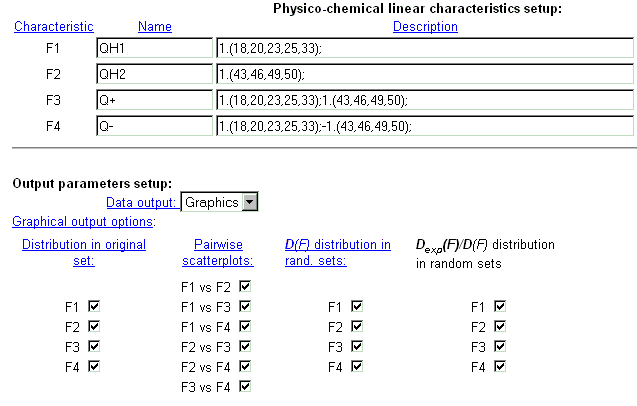
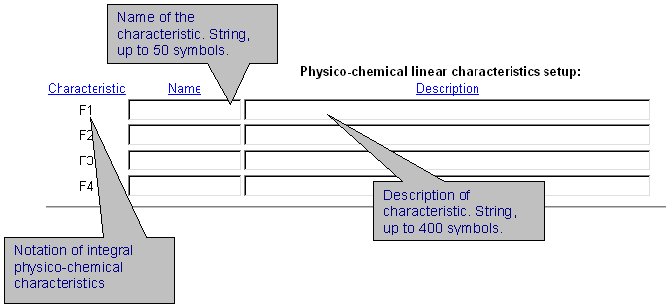
| It is possible to introduce up to 4 different integral protein characteristics that are related to the chosen physico-chemical property of residues. In the field 'Name' enter the name of characteristic. In the field 'Description', enter numerical coefficients for each protein position which influences the characteristics selected. Coefficients are ordered in a special format. See details here. |
 |
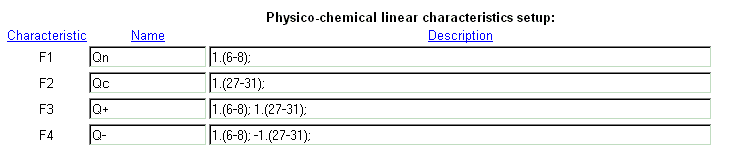
| Example of integral characteristics setup. |
 |
|
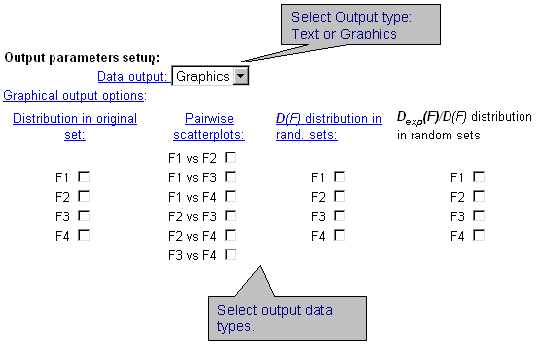
| Select the format of output data. The data could be displayed in a text form or as GIF-images. By clicking the check-boxes, it is possible to display information about distribution of characteristic values in original sample (first column), about dependence between two integral characteristics (second column), about distribution of characteristic variances in random samples (third column), about distribution of dispersion ratio in random samples (fourth column). |
 |
| Your actions with the
working window: Set up the following parameters:
|
|
| To RUN CRASP click the button 'Execute': |
 |
| Your actions with the working window:
|
| The result page will be displayed automatically. |