This is a pilot version of activation of the TRRD.
Programm SeqAnn (Version 0.993)
Activation of databases is a general term for extraction of the information that was not written in it in an explicit form. In particular, it is a knowledge discovery, employment of certain databases as a tool for accessing the other databases, etc.
The scheme of calculating of Scoren and Score(i) for the promoter XXXX. The cases when each recognized site makes impact into the integral Score are indicated by the arrow.
The basic idea of the proposed approach is the employment of the information on structure-function organization of promoters compiled in TRRD as a ready-made scenarios for their recognition. In the simplest case, it allows the search for the regions of similarity to a given promoter in an arbitrary sequence.
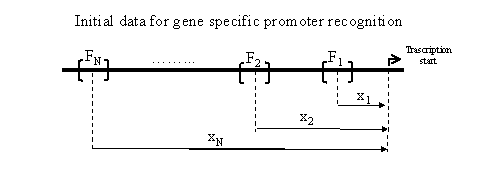
An information about promoter essential for its searching in the sequence under study is being extracted from TRRD. The information extracted is presented by the name of promoter, the length of promoter Lp and N sites of binding Fn, where for each nth binding site, its name, the position of the center xn and the length sn, are being extracted, 1<=n<=N.
On the basis of recognition of transcription factor binding sites, the system performs the search of the most likely transcription start in the nucleotide sequence S. For this purpose, a window W with the length Lp and the right boundary i is sliding along the sequence and the Score(i) estimating the probability of transcription site location at the point i is calculated. The window sequentially moves along the sequence and the similarity profile Score(i) is being constructed.
Let us consider the calculation of Score(i) in more details. The impact of nth site located at the ith position of the sequence into the promoter Score(i) value depends upon its distance from the current position i. The estimation of the Score(i) is performed according to direct search of coincidence with the footprint of the site under study. Besides, the fact could be taken into consideration that the binding sites could be located inside the definite interval along the sequence under consideration. For this purpose, for each nth binding site, an individual interval as a parameter could be introduced. In this case, the maximal value of the Scoren(i) on the interval xn±Dn is accounted for similarity profile Score(i) calculation.
Thus, the nucleotide sequence S={si} with the length Lp is analyzed to construct the set of N similarity profiles Scoren(i) for each nth binding site of this promoter:
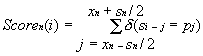 , (1)
, (1)
where ![]()
Equation 1 ascribes to the ith position of the sequence S the number {Scoren(i)} of coincidences of its region with the boundaries (i-sn/2, i+sn/2) with the region of the promoter considered with the boundaries (an,bn), that is, with the binding site of each nth transcription factor. Then the integral similarity profile {Score(i)} of this sequence and entire promoter is constructed:
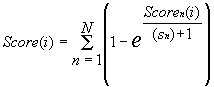 (2)
(2)
Equation 2 ascribes to the ith position of the sequence S the value of the similarity to the transcription start of the promoter P considered: the greater is the integral similarity of each of the considered nth binding sites of this promoter to this region of this sequence, the greater in the ascribed value.
The Scoren(i) is used to predict the potential transcription starts in the sequence S as follows. The mean value M and standard deviation s are calculated and used to find the region with the borders {c,d} within which the value Scoren(i) exceeds the threshold value M+3*s corresponding to the confidence interval s~0.01 of the Student's test with the number of degrees of freedom >>100. This region houses the maximal value Score(t), and the position t is predicted as a potential transcription start T of the sequences S. When K such regions {ck,dk} are found, K potential transcription starts {tk} is predicted (here 1<=k<=K).
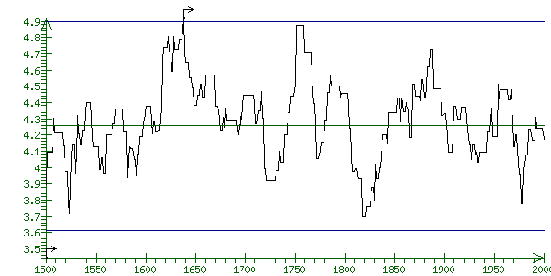
Fig 2. The element of the integral similarity profile Score(i) for recognition of the promoter extracted from the database TRRD according to ID=Hs:PBGD for the sequence extracted from the database EMBL according to AC=X73839 with the determined transcription start.
The system is available via the Internet at http://wwwmgs.bionet.nsc.ru/Programs/SeqAnn/.
Initial information from the TRRD database for the search for searching for promoter Hs:PBGD of porphobilinogen deaminase gene
Site |
Coordinates relative to the transcription start |
Site sequence |
Sp1 |
-198 |
TTCCCGCCCA |
Sp1 |
-186 |
AGGGAGGGAC |
NE-E1 |
199 |
AGATAA |
b3/c2 bs |
-185 |
GATAATGAA |
AP-1 |
-162 |
TGACTCAG |
CAC box |
-102 |
CACCC |
GATA-1 |
-73 |
TTATCT |
4 |
TCCTGGTTAC |
|
NF-E1 box |
44 |
CTATCG |
CAC box |
43 |
ACTATCGC |
The results of the application of equations 29 and 30 to a sequence extracted form EMBL by AC=X73839 (A.thaliana gene for hemC) and promoter of porphobilinogen deaminase extracted from TRRD by ID= Hs:PBGD are shown in Fig. 31. Note that the algorithm described above predicted in this sequence one potential transcription start at position 1638. According to the information contained in the field FT, this sequence has the transcription start at position 1603.
Thus, the central idea of this approach lies in the suggestion that any card of the TRRD database is a source of scenarios for searching for promoters of the same specificity in a newly sequenced nucleotide sequences. The user only should indicate the type of promoters (or an individual promoter) that is of interest to his studies.
The program that will develop this approach will include the interface with TRRD to allow the user to view the information it contains and select the sequences required for his analysis. The pilot release of the system SeqAnn (http://wwwmgs.bionet.nsc.ru/mgs/programs/seqann/) performs the search for the sites in a sequence during promoter recognition in the simplest form: by direct homology between each site contained in this promoter according to its description in TRRD and the sequence under study. In this case, TRRD is the source of information on not only a specific set of sites occurring in this promoter but on the relative location of these sites and, what is most essential, on their nucleotide sequences.
At the next stage, in search for the sites, this system will be used as a homology-based evaluation method, and further, as a more complicated recognition methods described above , which we are developing. The information on the activity value of the sites to be recognized will be employed. As it was already mentioned, the principle peculiarity of our approach is the automated discovery of the knowledge on structure-function organization of the sites and automated generation of the corresponding recognition programs. Consequently, a great number of complementary recognition programs will be developed for each type of functional sites and accumulated in the knowledge bases. This will provide a high potential of the system ACT_TTRD for generation of more complicated scenarios and increase in the accuracy of the regulatory region recognition.
Thus, the essence of the activation of the TRRD database within the frames of the approach suggested is its transformation from a passive source of reference information, as are all currently available databases, into an active information carrier, the key task of which is to generate scenarios for analysis of nucleotide sequences. The main stages of the recognition performance by this system will be: (1) automatic extraction of the information on a gene (promoter) and the functional sites contained in it; (2) selection of promoter to be recognized by the user; (3) extraction of the relevant programs for recognition of concrete functional sites; (4) generation of new recognition programs by the tools provided, if the available programs do not meet the user's requirements; (5) carrying out the recognition; (6) visualization of the results obtained; and (7) comparison of the results obtained with the initial information contained in the TRRD database. In addition, the user will have the possibility to modify the proposed active scenario either adding the sites (or another regulatory elements which are provided with the recognition programs), or removing the stages of recognition of certain sites.
![]()
This resource has been developed in Institute of Cytology and
Genetics. Novosibirsk, Russia
Authors: A.S. Frolov, S.V.
Lavryushev, D.A. Grigorovich
Contributors: D.G. Vorobiev, M.P. Ponomorenko
Leader: N.A.Kolchanov