PROF_PAT 1.3: Updated database
of patterns used to detect local similarities
A.G. Bachinsky1*, A.S. Frolov2,
A.N. Naumochkin1,
L.Ph. Nizolenko1, A.A. Yarigin1
1Theoretical
Department, Research Institute of Molecular Biology,
SRC VB 'Vector', Koltsovo, Novosibirsk region,
633159, Russia,
2Institute of
Cytology and Genetics SB RAS, Novosibirsk, 630090, Russia
* To whom reprint requests should be sent
Key words: protein families; patterns; motifs, similarity
search, database.
Running head: Database of patterns of protein families
ABSTRACT
Motivation.
When analysing novel protein sequences, it is now essential to
extend search strategies to include a range of 'secondary'
databases. Pattern databases have become vital tools for
identifying distant relationships in sequences, and hence for
predicting protein function and structure. The main drawback of
such methods is the relatively small representation of proteins
in trial samples at the time of their construction. Therefore a
negative result of an amino acid sequence comparison with such a
databank forces a researcher to search for similarities in the
original protein banks. We developed a database of patterns
constructed for groups of related proteins with maximum
representation of amino acid sequences of SWISS-PROT in the
groups.
Results.
Software tools and a new method have been
designed to construct patterns of protein families.
By using such method, a new version of databank of
protein family patterns, PROF_PAT 1.3, is produced. This bank is
based on SWISS-PROT (rl.38) and TrEMBL (rl.11), and contains
patterns of more than 13,000 groups of related proteins in a
format similar to that of the PROSITE. Motifs of patterns, which
had the minimum level of probability to be found in random
sequences, were selected. Flexible fast search program
accompanies the bank. The researcher can specify a similarity
matrix (the type PAM, BLOSUM and other). Variable levels of
similarity can be set (permitting search strategies ranging from
exact matches to increasing levels of "fuzziness").
Availability.
The Internet address for comparing sequences with the bank is:
http://wwwmgs.bionet.nsc.ru/mgs/programs/prof_pat/. The local
version of the bank and search programs (approximately 50 Mb) is
available via ftp:
ftp://ftp.bionet.nsc.ru/pub/biology/vector/prof_pat/, and
ftp://ftp.ebi.ac.uk/pub/databases/prof_pat/. Another appropriate
way for its external use is to mail amino acid sequences to
bachin@vector.nsc.ru for comparison with PROF_PAT 1.3.
Contact.
bachin@vector.nsc.ru
INTRODUCTION
Up to now, the main method of suggesting possible functions of
the newly deciphered amino acid sequences has been to search them
for similarity with sequences available in protein banks such as
PIR (Barker et al., 1999), SWISS-PROT (Bairoch and Apweiler,
1999) and others. As these banks grow larger, such comparisons
become more promising but at the same time more time-consuming.
In addition, in the case of distant proteins the search for
global similarity of complete sequences may fail to show a
positive result, because the conservative blocks responsible for
their special functions may prove to be relatively short and
scattered all over the sequence. This may be why a number of
works appeared in the last few years, aimed at the selection of
sites in groups of related proteins. These sites are
representative of a protein family as a whole, and both identify
new proteins and refine structural and functional properties of
those already known. Such databases as PROSITE (Hofmann, et al.,
1999), BLOCKS (Henikoff and Henikoff, 1991, Henikoff, et al.,
1999), PRINTS (Attwood et al., 1999) are among the most well
known and accessible via Internet. There is also a number of
other similar databases i.e. PFAM (Bateman, et al., 1999), SBASE
(Murvai, et al., 1999), IDENTIFY (Nevill-Manning, et al. 1998).
The advantages of such databases, representing protein
families, arise from comparing an amino acid sequence with a
relatively small pattern as a representative of a protein family,
and not with a set of long sequences of this family. Furthermore,
comparison algorithms could be simplified because there is no
need of sequence alignment, which also speeds up the process. The
level of noise (random similarity) is lower, because comparison
is usually made with patterns that are already characterised and
represent conservative intervals of positions. This appears most
clearly when a family is represented by a few relatively short
pattern motifs.
However the common shortcoming found for the most part in
secondary databases, is the fact that they contain a limited
number of patterns (profiles, motifs, alignments or other
representations of protein families). In general, PROSITE
contains well-defined manually constructed patterns.
BLOCKS and PRINTS in the beginning were in some extent expansions
of PROSITE with subsequent developments. In turn, IDENTIFY is
based on BLOCKS and PRINTS.
We have devoted our efforts to develop a technique and
construct patterns for the greatest possible number of proteins
belonging to the SWISS-PROT+TrEMBL (Bachinsky et al., 1996,
1997). We are convinced that if a secondary bank is not really
representative, it would not be widely used. It is because of
negative results in the comparison of a sequence with this bank
that the user is forced to consult other banks or make direct
comparisons of the sequence with large banks of sequences.
SYSTEMS AND METHODS
Programs for using patterns were written in Microsoft Visual
C++ 5.0. for IBM compatible computers running Windows 95 or
Windows NT. Amino acid sequences were taken from release 38 of
the bank SWISS-PROT and release 11 of TrEMBL (1999). The
selection of related proteins was made using modified FASTA 2.0.
algorithm (Pearson, 1994). Multiple alignments of the amino acid
sequences were performed by means of CLUSTALV program (Higgins,
et al., 1992) and the default set of parameters.
ALGORITHMS
The selection and concurrent alignment of related protein
groups. All full-length sequences of prototype
banks that were over 30 amino acids in length were combined in
one file. In order to select groups of related proteins, a
special program was written based on FASTA 2.0 (Pearson, 1994).
The first sequence of the file was compared with all other
sequences. The sequences were regarded as similar if score/ln(l1)/ ln(l2)*ln100*ln100
> 80. The sequences similar in the sense of FASTA form a
primary set of related proteins. Then the next sequence not
included in a group of related proteins was compared with all
other proteins, and so on.
It is known that in the case of distant protein homology many
algorithms of multiple alignment lead to doubtful results
strongly dependent on parameters such as, insertion penalties,
similarity matrix, etc. To provide quality alignment, the
pairwise similarity level was prescribed 30% or more, for it is
known that at smaller similarity global alignment often makes no
sense (Vogt, et al., 1995; Patthy, 1987). Pairwise similarity of
the proteins belonging to a set was assessed by the program
CLUSTALV (Higgins et al., 1992). Then, if not all pairs of
proteins had 30% similarity, the set was divided into subsets so
that all pairwise similarities were at least 30%. Thus, more than
13,000 subsets or groups were obtained, containing more than 100
000 sequences.
Proteins of every subset were aligned together. The files
containing aligned sequences were supplemented with two fields:
DE (description(s) of proteins forming the group), and KW ( key
words; mainly the union of values of field KW for proteins
falling into the set). Patterns were constructed based
on such aligned families.
The construction of patterns of protein families. We
will regard the combination of motifs that represent relatively
conservative intervals of positions of aligned proteins of the
family as a pattern of a family of related proteins.
Let there be an interval of positions of an aligned family of
related proteins of length n. Ai
denotes a subset of amino acids of 20 standard ones, located in a
position i of the interval. An amino acid sequence of
length n will be considered to belong to the given
interval (motif), if for every position ai belongs to Ai,
where ai is the
amino acid located in position i of the sequence. To every
position i of a group of aligned proteins the value 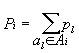 that is the frequency of
occurrence in proteins one of amino acids from Ai located in this position
may be ascribed. Here pl
is the frequency of occurrence in proteins of amino acid al, located in this position. Qj - the product of the values
Pi for positions
falling into interval of positions j and may, accordingly,
be the characteristic of this interval. The value Qj constitutes an assessment
of probability that a random amino acid sequence of length n
will belong to interval j. The smaller Qj is, the smaller is the
probability that, in a protein (a random sequence) not related to
the given group, a fragment belonging to the interval can be
found. Thus the smaller Qj
is, the greater is its ability to differentiate fragments of
proteins of the trial sample from fragments of other proteins,
i.e. its specificity.
that is the frequency of
occurrence in proteins one of amino acids from Ai located in this position
may be ascribed. Here pl
is the frequency of occurrence in proteins of amino acid al, located in this position. Qj - the product of the values
Pi for positions
falling into interval of positions j and may, accordingly,
be the characteristic of this interval. The value Qj constitutes an assessment
of probability that a random amino acid sequence of length n
will belong to interval j. The smaller Qj is, the smaller is the
probability that, in a protein (a random sequence) not related to
the given group, a fragment belonging to the interval can be
found. Thus the smaller Qj
is, the greater is its ability to differentiate fragments of
proteins of the trial sample from fragments of other proteins,
i.e. its specificity.
Some positions may be marked as ‘passive’ or
‘non-meaningful’ (in comparison with patterns of
PROSITE). They do not influence the definition of value Qj, and comparison of these
positions is not implemented in pattern analysis (any amino acids
are acceptable). In our case all positions that had more than 4
amino acids and/or the total frequency of amino acid occurrence
more than 0.2, were regarded as passive. Having analysed the
structure of the bank PROSITE, we found out that about 80% of the
bank’s patterns fall into the following boundaries: up to 10
‘active’ positions and a total length of no more than
20 positions. We also used these limits when choosing pattern
motifs.
Having set some critical value of the Q’, for a
series of chosen proteins one can get a set of motifs R(Q’,
l1, l2),
the borders of which are limited to l1,
l2. Characteristics of
motif j are the value Qj
that determines its specificity, and length n.
To ensure an effective realisation of comparison algorithm
between sequences and patterns, we demand that every motif should
contain an ungapped section of no less than four active
positions.
The motifs of patterns are represented by ambiguous words of
the type:
K-[D,E] - F - [I,V] - C - X - [A, S, T] - X - [M, N, D]. Thus,
an initial pattern of a protein family is an ordered combination
of non-overlapping motifs of the type r:A1-A2-A3-...-An. Here r is position
number of an aligned group of proteins (the trial sample), where
the motif begins, Ai
is a set of amino acids, located in r+i-1 position of the
trial sample. For a passive position Ai
= X: any amino acid is acceptable. The number of motifs
does not exceed 5 per 100 positions.
Each pattern is compared with all sequences of the initial
file. If not less than 60% of motifs of a pattern reveal
similarity with a sequence that is not included in other
families, an attempt to include this sequence into the trial
sample is made. If any motif is non-specific (i.e. it matches
many sequences, but other motifs of the pattern do not match
them) the motif is excluded from the pattern.
COMPARISON OF AMINO ACID SEQUENCES WITH PATTERNS
The searches for exact matching between fragments and
pattern motifs of amino acid sequences. The main algorithm
for comparing an amino acid sequence with the pattern database
uses the modification of finite automaton of Aho-Corasic (Aho and
Corasic, 1975), constructed based on a set of samples, which are
to be searched for in the input text. The automaton is presented
as an oriented tree-like graph, where nodes are states of the
automaton and arcs are admissible transitions from some states to
the others, marked with symbols from the alphabet S of the
amino acid designation. The automaton works in cycles. In every
cycle one more symbol of a text is read, which determines the
transition of the automaton from the current state into a new
one. The behaviour of the automaton is characterised by three
functions: function of transitions G(s,a);
rejections’ function F(s) and output function O(s).
The values of these functions are calculated once when
constructing the automaton based on a given set of samples. In
Figure 1. the functions of the automaton constructed on the set
of samples R={r1, r2, r3,
r4, r5}
= {HE, SHE, HIS, HER, HERS} are illustrated.
The function of transitions s'=G(s,a) determines into
what state s’ the automaton passes from a current
state s, if the input symbol is 'a'. When there is
no admissible transition, a situation called
‘rejection’ arises which indicates that the comparison
has failed.
The values of the rejections’ function F(s) are
calculated in the situations, when G(s,a) =
‘rejection’. In this case a backward move does not
occur through the text to the beginning of another fragment of
the sequence (exit to the initial state of the automaton). A new
sample is being examined from the break site of the previous one.
It provides linear dependence of the search time on the length of
the query sequence. F(s) indicates into what state the
automaton passes from a current state s, if the next
symbol of the text does not coincide with a label of any of the
arcs, which go out from s. The transitions ‘upon
rejections’ are the ones that guarantee the returnless
manner of text scanning.
The output function O(s) indicates the list of motifs,
represented by a sample (as a sequence of arc labels on the way
from initial state ‘0’ into ‘s’), a
successful search for which is realised as the automaton passes
into state s.
Fig. 1. Illustrations of the functions of
the Aho-Corasic automaton constructed on the set of samples
R={r1, r2, r3, r4, r5} = {HE, SHE, HIS, HER, HERS}. a) Graph representation
of transition function G(s,a). b) Rejections’
function F(s). c) Output function O(s). d)
Transition of Automaton from state to state if the input text is
"ushers"
When constructing the automaton, in every motif four
neighbouring positions are chosen (the core of a motif), having
minimum value of the product Pi
and containing no passive positions. Then this core is converted
into exactly determined words of length 4 that act as samples in
constructing automaton. If coincidence of a current fragment of
an input sequence and one of the automaton samples is observed,
comparison is performed (up to the first non coincidence) of all
the other motif positions from the list of the output function,
and the corresponding fragments of the sequence (the stage of
extending the core). According to the results of this stage, the
final decision is made on whether there is similarity or not.
The search for distant similarity. To reveal a distant
similarity, the algorithm of comparison is modified. The user
specifies the matrix of similarity of amino acid residues [e.g.,
using the one from families PAM (Dayhoff, 1978), BLOSUM, etc.]
and D - the level of similarity within the limits of
motif. For all states of the automaton, the function of rejection
is set to zero. Besides, a sequence as a whole does not input to
the automaton, but specially processed words. Preparing words,
additional to the initial fragment and the search process are as
follows:
A. For every fragment of the input sequence of length 4 the
value So is calculated - the
sum of values of diagonal elements of the matrix of similarity
for amino acids of the fragment. Then the value So = S04
+ 6*Sm is calculated, where  is average value of diagonal elements sii of the scoring matrix, pl is the frequency of occurrence in
proteins of amino acid i. The coefficient 6 is chosen
because of the length 10 is the most usual for motifs.
is average value of diagonal elements sii of the scoring matrix, pl is the frequency of occurrence in
proteins of amino acid i. The coefficient 6 is chosen
because of the length 10 is the most usual for motifs.
B. Supplementary ‘similar’ words are constructed.
For each possible word of length 4, the sum of elements of the
similarity matrix 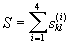 is calculated. Here k and l are indices of amino
acids of the initial fragment and the supplementary word in
corresponding positions i,
is calculated. Here k and l are indices of amino
acids of the initial fragment and the supplementary word in
corresponding positions i,  is an element of the similarity matrix. If S
does not differ from the sum of diagonal elements for the
corresponding amino acids of the fragment by a value greater than
(100-D)*So/100, where D (%) is the point of similarity,
specified by the user, the word is accepted.
is an element of the similarity matrix. If S
does not differ from the sum of diagonal elements for the
corresponding amino acids of the fragment by a value greater than
(100-D)*So/100, where D (%) is the point of similarity,
specified by the user, the word is accepted.
C. All these words are passed to the entry of the automaton
consecutively. In the case of coincidence with the sample, the
following values are calculated: 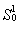 the sum of diagonal values of the
similarity matrix for amino acids of the fragment, corresponding
to active positions of the motif, and
the sum of diagonal values of the
similarity matrix for amino acids of the fragment, corresponding
to active positions of the motif, and 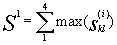 similarity of the motif to the fragment.
Here k is index of amino acid, located in the position of
the fragment, max(
similarity of the motif to the fragment.
Here k is index of amino acid, located in the position of
the fragment, max( )
is the maximum value of similarity for amino acid k of the
fragment and amino acids l, located in position i
of the motif. If Si>
*D/100, then the
decision is made about the existence of the prescribed degree of
similarity.
)
is the maximum value of similarity for amino acid k of the
fragment and amino acids l, located in position i
of the motif. If Si>
*D/100, then the
decision is made about the existence of the prescribed degree of
similarity.
The comparison of patterns with the parent banks. To
examine the recognising ability of the patterns and exclude
certain motifs, which are non-specific for a given family, all
patterns were compared with all the proteins of the
SWISS-PROT+TrEMBL. In the routine comparison between patterns and
the banks, only exact similarities of two or more motifs per a
pattern were registered, i.e. the cases when fragments of amino
acid sequences belong to the motifs. The comparisons made the
inclusion of hundreds of sequences into trial samples possible.
The similarity is regarded as ‘positive’ one, if at
least one of the two following conditions is met.
1. Query sequence belongs to the trial sample.
2. All words of one of the DE fields of the pattern (the names of
the proteins forming the family) are present in the field DE
(protein name) of the sequence.
The last condition needs some clarification. Proteins are
considered to be related to the trial sample proteins, if they
have the same name that the trial sample has, possibly
supplemented with words HYPOTHETICAL, PROBABLE, POSSIBLE,
PRECURSOR, CHAIN, etc., descriptions of localisation of sequences
(MITOCHONDRION, CHLOROPLAST, PLASMID), its composition in the
case of polyproteins, a note concerning the type of the chain,
etc.
The similarity is considered ‘conditionally
positive’ (UNKNOWN), if at least one of the DE or KW words
of the pattern coincides with one of the words determined in
fields DE and/or KW of the sequence. Thus, proteins are defined
as conditionally related if they possess some common function
(e.g., hydrolases, dehydrogenases, oxidoreductases, etc.) or some
specific features of their structure (for instance, transmembrane
segments). All other cases of similarity are regarded as false
positive. As a result of comparison with the bank, a pattern bank
entry is created, similar in its structure to entries of PROSITE.
The example of such entry is given in Figure 2. Descriptions of
the fields are given in Table 1.
ID 00004;
DT Thu Aug 5
14:35:08 1999
DE
4-HYDROXYPHENYLPYRUVATE DIOXYGENASE (EC 1.13.11.27) (4HPPD)
KW
OXIDOREDUCTASE; DIOXYGENASE; IRON; ACETYLATION.
PR P32754,
P49429, Q02110, Q22633, Q27203, Q00415,
PR O42764,
Q53586;
RE
SWISS-PROTEIN(rl.37)+TREMBL(rl.8)
AC 00004_1
(47-56)
PA
[FW]-[AVWY]-V-G-N-A-K-Q-[AV]-A
FR frequency
= 4.02065e-12
DR P32754, T;
P49429, T; Q02110, T; Q22633, T; Q27203, T;
DR Q00415, T;
O42764, T; Q53586, T;
NR
/TOTAL=8(8); /POSITIVE=8(8); /UNKNOWN=0(0); /FALSE_POS=0(0);
NR
/FALSE_NEG=0;
*/
...
AC 00004_12
(391-400)
PA
D-R-P-T-[LV]-F-[FILY]-E-[IV]-I
FR frequency
= 4.6197e-12
DR P32754, T;
P49429, T; Q02110, T; Q22633, T; Q27203, T;
DR O23920, T;
Q00415, T; O42764, T; Q53586, T; Q18347, ?;
NR
/TOTAL=10(10); /POSITIVE=9(9); /UNKNOWN=1(1); /FALSE_POS=0(0);
NR
/FALSE_NEG=0;
*/
//
Fig. 2. An example of the pattern bank
entry.
Table 1.
Descriptions of fields of PROF_PAT 1.3. entries
Identificator
of a field
|
Value of
the field
|
ID
|
Unique identificator of a
pattern (entry) |
DT
|
Date of creation/edition |
DE
|
Description of the pattern,
usually - descriptions (DE) of sequences of the trial
sample |
KW
|
Key words |
PR
|
The set of ACs of sequences
of the trial sample |
RE
|
Release of parent bank |
AC
|
Identifier and margins of
the motif of the pattern |
PA
|
Motif of the pattern |
FR
|
Occurrence frequency of
matching of the motif to random fragment of amino acid
sequence |
DR
|
The
set of ACs of amino acid sequences of parent bank that
match the motif: Ò - true, ? – unknown, F - false
positive |
NR
|
The
statistics of matches of the motif to amino acid
sequences of the parent bank: Õ(Ó), where Õ is the
total number of matches, Ó - the number of
sequences where matches have been revealed |
*/
|
The end of the motif |
//
|
The end of the pattern |
DISCUSSION
In the first version of PROF_PAT, patterns were selected in
accordance with the principle of small variability of several
physicochemical properties of amino acids (Bachinsky, et al.,
1997). However, the use of this bank in actual practice revealed
its shortcomings. Its large size and the time-consuming procedure
of comparison compelled us to modify its construction principles
to a certain extant.
In version 1.3. presented here, the total number of motifs in
more than 13,000 patterns is over 200,000 with specificities
varying from one expected false positive prediction in 108
tests and higher. The total combined length of patterns is about
2 million positions.
To find a distant similarity, a very fast flexible comparison
procedure is employed, using the modified algorithm of
Aho-Corasic (Aho and Corasic, 1975), various matrices of
similarity/distance for amino acid residues, the predetermined
grade of similarity between a fragment of an amino acid sequence
and a pattern motif.
The results of comparison of patterns with all sequences of
release 38 of the SWISS-PROT and release 11 of TrEMBL are given
in Table 2. Patterns identify nearly 130 000 of amino acid
sequences as having shown ‘positive’ or
‘conditionally-positive’ similarity. In the latter
case, the similar sequences, not included into the trial samples,
are usually identified.
Table 2.
Results of comparisons of sequences of parent banks with
patterns of PROF_PAT 1.3.
| Number of sequences with
positive matches |
127,634
|
| Number of sequences with
conditionally-positive matches |
2,317
|
| Number of sequences with
false-positive matches |
1238
|
| Number of sequences of trial
samples that do not match one or more motifs of the
patterns |
2177
|
| Number of sequences of trial
samples that do not match any motif of the patterns |
0
|
Almost all sequences of the trial samples are recognised by
all motifs of the corresponding patterns. Certain violations of
this rule are due only to the presence of non-standard symbols in
the particular sequences of the trial samples that have fallen
into the intervals of positions represented by pattern motifs.
A number of cases of false-positive similarity may be divided
into two classes. Sometimes it is a really chance similarity.
However, sometimes two or more pattern motifs show similarity to
the fragments of a certain sequence; the order of the
fragments’ locations often correspond to that of the
motifs’ locations, which increases even more the certainty
that the similarity is not random. In most cases, false-positive
similarity is revealed with sequences described only as products
of some genes, and this information is not included into
descriptions of patterns (see some examples in Table 3.).
Table 3.
Some cases of false-positive similarity have
been revealed, when patterns of PROF_PAT 1.1. bank were searched
for SWISS-PROT+TrEMBL bank
Descriptions of protein families (patterns)
|
Entry names of related sequences
|
Number of similar motifs/number. of motifs in
the pattern
|
Descriptions of sequences
|
Comments
|
| SERINE
THREONINE KINASE |
Q26345
|
5/15
|
FU (FUM1)=SEGMENT POLARITY GENE FUSED {28-BP
DELETION}.
|
The sequence matches motifs of the N-end of the
pattern
|
FLORAL
HOMEOTIC PROTEIN AGL
AGAMOUS PROTEIN
MADS BOX PROTEIN
DAL2 PROTEIN |
Q41876
|
5/9
|
ZAG1.
|
|
| COLLAGEN |
Q20927
|
4/7
|
F57B7.3.
|
|
RNA-DIRECTED
RNA POLYMERASE
(EC 2.7.7.48)
READTHROUGH PROTEIN REPLICASE
REPLICATION-ASSOCIATED PROTEIN |
Q84126
Q88598
|
20/66
20/66
|
52KDA UNKNOWN IN FUNCTION PROTEIN
54 KDA PROTEIN
|
The sequences match motifs of the C-end of the
pattern
|
| ZINC-FINGER
PROTEIN |
Q60980
|
6/19
|
BKLF.
|
|
| PHOSPHOLIPASE |
Q63693
|
29/30
|
PHODPHOLIPASE C DELTA4.
|
An error in protein DE
|
ATP SYNTHASE
A CHAIN (EC 3.6.1.34) (PROTEIN 6)
ATPASE
ATP SYNTHASE SUBUNIT 6 |
Q35294
|
4/12
|
URF-RMC
|
The sequence matches motifs of the N-end of the
pattern
|
| PROTEIN B15 |
VC16_VACCC
|
4/8
|
PROTEIN C16/B22.
|
|
CANAVALIN
BETA-CONGLYCININ |
Q39816
|
11/23
|
7S STORAGE PROTEIN ALPHA SUBUNIT.
|
The sequence matches motifs of the C-end of the
pattern
|
TRANSPOSASE
MARINER PROTEIN |
YKC6_CAEEL
|
5/10
|
HYPOTHETICAL 29.3 KD PROTEIN B0280.6 IN
CHROMOSOME III.
|
The sequence matches motifs of the C-end of the
pattern
|
| MASC PROTEIN |
Q50586
|
4/15
|
HYPOTHETICAL 63.1 KD PROTEIN.
|
|
| MUCIN |
Q22902
|
9/11
|
COSMID C16D9.
|
The sequence matches motifs of the N-end of the
pattern
|
NUCLEOLAR
PROTEIN
NUCLEOPHOSMIN-RETINOIC ACID RECEPTOR
ALPHA FUSION PROTEIN |
Q14115
|
7/14
|
P80 PROTEIN.
|
The sequence matches motifs of the N-end of the
pattern
|
| PROTEIN
KINASE |
Q24096
Q24590
|
4/18
4/18
|
LATS.
TUMOR SUPPRESSOR.
|
RNA-DIRECTED
RNA POLYMERASE (EC 2.7.7.48)
REPLICASE |
Q65014
Q83423
Q83426
|
8/29
5/29
5/29
|
PROTEIN OF 33 KDA.
PROTEIN 29.
29K PROTEIN.
|
The sequences match motifs of the N-end of the
pattern
|
SPLICING
FACTOR
RNA BINDING PROTEIN 1 |
Q62093
|
4/5
|
PR264/SC35.
|
The sequence matches motifs of the N-end of the
pattern
|
SUPPRESSOR OF
FORKED PROTEIN
CLEAVAGE STIMULATION FACTOR |
Q24539
|
13/34
|
39 KD PROTEIN.
|
The sequence matches motifs of the N-end of the
pattern
|
| TRANSCRIPTION
FACTOR |
Q18694
|
3/9
|
C47G2.2.
|
|
TRANSPOSASE
TNIA |
Q47380
|
9/26
|
INVASIN
|
The sequence doesn't match the INVASIN pattern.
The membership to this class of proteins is doubtful
|
| TRANSALDOLASE
(EC 2.2.1.2) |
Q49705
Q49698
|
6/18
6/18
|
B1496_F2_65.
B1496_F1_27.
|
The sequences match motifs of the N-end (Q49705)
and C-end (Q49698) of the pattern
|
| TRANSPOSASE |
ADPR_LACLA
|
5/12
|
ATP-DEPENDENT PROTEASE (EC 3.4.21.-).
|
The sequence doesn't match the patterns of
ATP-dependent proteases
|
| PROBABLE
TRANSPOSASE FOR INSERTION SEQUENCE ELEMENT IS701 |
Q55975
Q55976
|
5/11
5/11
|
HYPOTHETICAL 16.4 KD PROTEIN
HYPOTHETICAL 13.5 KD
PROTEIN.
|
The sequences match motifs of the N-end (Q55975)
and C-end Q55976) of the pattern
|
RECEPTOR-LIKE
TYROSINE-PROTEIN KINASE
(EC 2.7.1.112) |
Q23677
|
12/15
|
ZK938.5.
|
The sequence matches motifs of the C-end of the
pattern
|
| CAPSID
PROTEIN |
COA3_AAV2
|
13/32
|
PROBABLE COAT PROTEIN 3.
|
|
| PROTEIN VP2 |
Q84387
|
4/23
|
HYPOTHETICAL 7.9 KD PROTEIN.
|
It is a C-fragment of VP2 protein
|
| ANAEROBIC
RIBONUCLEOSIDE-TRIPHOSPHATE REDUCTASE (EC 1.17.4.2) |
Q38428
|
19/28
|
ORF (55.11).
|
The virus genome contains a C-fragment of
bacterial protein
|
| DIHYDROPYRIMIDINASE |
Q21773
|
8/20
|
R06C7.3
|
|
| REVERSE
TRANSCRIPTASE |
Q63305
|
15/57
|
LONG INTERSPERSED REPETITIVE DNA CONTAINING 7
ORF'S.
|
It is a C-fragment of transcriptase
|
DEHYDROGENASE
DIOXYGENASE |
Q52459
|
9/9
|
THE FIRST START CODON IN THE ORF IS FOUND AT
POSITION 2466.
|
Besides of the sequence matches all motifs of
the pattern, it hasn't 30%similarity with all proteins of
the trial sample
|
All patterns were searched in proteins of more recent version
of SWISS-PROT that were absent in parent version (file
new_seq.dat on the EBI server, 21, September 1999). This
comparison was made with the purpose of testing PROF_PAT for
sensitivity and to work through the technology of the bank
update. The first 500 complete long sequences (i.e. 30 or more
amino acid residues in length) that do not contain words
'PUTATIVE' or ‘HYPOTHETICAL’ were selected for testing.
Comparison was carried out at level of similarity 80 %. Matrix
250PAM was used. 73 sequences were not identified (no significant
matchings). Two sequences were identified by patterns that have
DE fields different from DE fields of the sequences. There were
425 sequences identified as ‘positive’. Of these 500
sequences 285 contain links to PROSITE patterns, and 245
sequences contain links to PFAM. From 1480 new sequences of
TrEMBL described as ORFs, 419 were undoubtedly identified. It
should be mentioned that some of these matchings refer the
sequences to patterns or protein groups described as ORFs or
HYPOTHETICAL PROTEINS also. IDENTIFY assigns biological functions
to 25-30% of all proteins encoded by the Saccharomyces cerevisiae
genome and by several bacterial genomes (Nevill-Manning, et al.
1998). Thus, the results of comparisons for these two banks are
alike.
Some other comparisons of the PROF_PAT and other secondary
banks are presented in Table 4. They show that PROF_PAT exceeds
the most popular banks PROSITE, PRINTS, and BLOCKS under such
index as number of patterns and motifs. The more pattern motifs
show similarity to the sites of a query sequence, the higher
would be the likelihood that the amino acid sequence is related
to proteins of the trial sample (Henikoff and Henikoff, 1991),
especially if the order of the motifs coincides with that of the
sample proteins. We can not make direct bulk comparisons of
PROF_PAT with other banks because other banks (with the exception
of PRINTS) do not provide search of many sequences in one
connection.
Thus, we have constructed a bank of patterns for protein
families, representing about two-thirds of the full-length
protein sequences of the bank SWISS-PROT release 38 and TrEMBL
release 11. The fast flexible search program for close and
distant similarity provides comparisons of amino acid sequences
of interest with the bank of patterns in the interactive mode.
The PROF_PAT technology update has been developed and tested, so
the new versions of PROF_PAT will be created following each new
versions of SWISS-PROT+TrEMBL.
Table 4.
Some characteristics of secondary banks in
comparison with PROF_PAT.
| The name of the bank its
release/parent banks |
Number of patterns (entries) |
Number of motifs |
The source
of data
|
PROF_PAT 1.3.
August 1999 / SWISS-PROT 38 + TrEMBL 11 |
>13,000
|
>200,000
|
http://wwwmgs.bionet.nsc.ru/programs/prof_pat |
| PROSITE 15, July 1998 |
1,031
|
1,366
|
http://www.expasy.ch/prosite/ |
| PFAM 4.0, May 1999 /
SWISS-PROT 37 + TrEMBL 9 |
1,465
|
|
http://www.sanger.ac.uk/Pfam |
| PRINTS 22.0, March 1999 |
1,100
|
6,510
|
http://www.biochem.ucl.ac.uk/bsm/dbbrowser |
| BLOCKS 11.0, July 1998 |
994
|
4,034
|
http://www.blocks.fhcrc.org/ |
| SBASE 6.0 |
1,037
|
2,459
|
http://base.icgeb.trieste.it/sbase |
| PIMA |
22,416
|
|
Ladunga I., et al. (1997) |
| IDENTIFY, 1998
/PRINTS+BLOCKS |
7,000
|
50,000
|
http://motif.stanford.edu/identify |
REFERENCES
Aho,A.V. and Corasic,M.J. (1975) Efficient
String Matching: An Aid to Bibliographic Search. Commun. ACM, 18,
333-340
Attwood,T.K., et al. (1999) PRINTS
prepares for the new millennium.. Nucleic Acids Res., 27,
220-225.
Bachinsky,A.G., et al. (1996) A new
release of a bank protein family patterns PROF_PAT 1.0.: A
technology of construction and programs of fast search. Molecular
Biology (Russian), 30, 1409-1419.
Bachinsky,A.G. et al. (1997) A bank of
protein family patterns for rapid identification of possible
functions of amino acid sequences. Comput. Applic. Biosci.,
13, 115-122.
Bairoch,A. and Apweiler,R. (1999) The
SWISS-PROT protein sequence data bank and its supplement TrEMBL
in 1999. Nucl. Acids Res. 27, 49-54.
Barker,W.C., et al. (1999) The
PIR-International Protein Sequence Database. Nucleic Acids
Res., 27, 39-43.
Bateman,A., et al. (1999) Pfam 3.1: 1313
multiple alignments and profile HMMs match the majority of
proteins Nucl. Acids Res., 27, 260-262.
Dayhoff, M.O., Eck,R.V. and Park C.M. (1972) A
Model of Evolutionary Change in Proteins. In: Dayhoff, M.O. (ed)
Atlas of Protein Sequence and Structure, Silver Spring, MD:
National Biomedical Research Foundation. 5, 89-99.
Henikoff,S. and Henikoff,J.G. (1991) Automated
assembly of protein blocks for database searching. Nucl. Acids
Res., 19, 6565-6572.
Henikoff,J.G., Henikoff,S. and Pietrokovski,S.,
(1999) New features of the Blocks Database servers. Nucl.
Acids Res., 27, 226-228.
Higgins,D.G., Bleasby,A.G. and Fuch, R. (1992)
CLUSTAL V: Improved software for multiple sequence alignment. Comput.
Applic. Biosci., 8, 189-191.
Hofmann, K., et al. (1999) The PROSITE
database, its status in 1999; Nucleic Acids Res., 27,
215-219.
Ladunga,I., Wiese,B.A. and Smith R.F. (1996)
FASTA-SWAP and FASTA-PAT: Pattern database searches using
combination of aligned amino acids, and a novel scoring theory. J.
Mol. Biol., 259, 840-854.
Murvai,J., et al. (1999) The SBASE
protein domain library, release 6.0: a collection of annotated
protein sequence segments. Nucleic Acids Res., 27,
257-259.
Nevill-Manning, C.G., Wu, T.D. and Brutlag,
D.L. (1998) Highly specific protein sequence motifs for genome
analysis. Proc.Natl.Acad.Sci., 95, 5865-5871.
Patthy,L. (1987) Detecting homology of
distantly related proteins with consensus sequences. J. Mol.
Biol., 198, 567-577.
Pearson,W.R. (1994) Using the FASTA program to
search protein and DNA sequence databases. in Griffin A.M.,
Griffin H.G., (eds) Methods in Molecular Biology. Computer
analysis of sequence data. Part 1. Humana Press,
Totova. 24, pp.307-331.
Vogt,G., Etzold,T. and Argos,P. (1995) An
assessment of amino acid exchange matrices in aligning protein
sequences: The twilight zone revisited. J. Mol. Biol., 249,
816-831.
Wallace,J.C. and Henikoff,S (1992) PATMAT: a
searching and extraction program for sequence, pattern and block
queries and databases. Comput. Applic. Biosci., 46,
567-577.