MiRNA prediction by sequence similarity
Program description: The tool automatically identifies close miRNA homologs (the maximum difference is 2 symbols) in an arbitrary sequence based on a defined query miRNA or on all known miRNAs for a particular organism (miRBase.org, release 21.0).
Biological task that could be solved: Identification of novel homologous miRNAs.
Data input: Into the text-box (shown by the red arrow #1 in Fig.1), enter or insert from the clipboard a genomic sequence to be analyzed. You may input this sequence in the fasta format or in a plain text format without the comment line. The size limit is 10 000 symbols. Use the alphabets ATGC(atgc) or AUGC(augc), any other symbol in the nucleotide sequence will give an error. Line foldings and spaces are ignored.
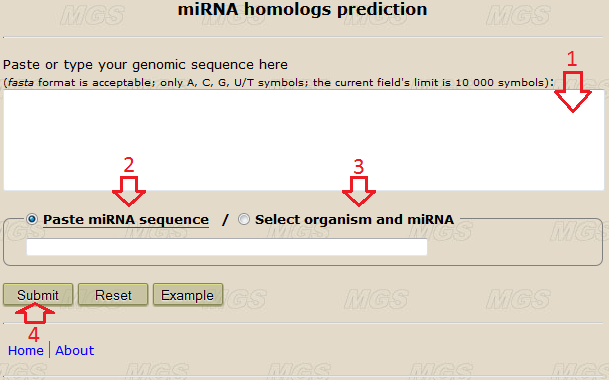
Figure 1. The main program window.
Program options: You may input one miRNA sequence in the text-field (red arrow #2 in Fig.1), or select an organism and a desired miRNA (or all miRNAs) from drop-down lists (red arrow #3 in Fig.1, you need to have JavaScript enabled in your browser). Use the alphabets ATGC(atgc) or AUGC(augc), any other symbol will give an error.
Program execution: Start the program by clicking the button "Submit" (red arrow #4).
Data output: Program execution will bring up the resulting window with the data output. The result includes the following (fig.2):
• The comment line from the text-box or the ID of the query miRNA and the name of the organism (if defined, red arrow #1);
• The sequence of the query miRNA (red arrow #1, marker "Rev." means an inverted miRNA sequence);
• The sequences of the predicted homologs and their positions in the query genomic sequence (red arrow #2).
Example: As an example, we search homologs for the sequence "UGAGGUAGUAGGUUGUAUAGUU" in the pre-miRNA "hsa-let-7a-1". The output is represented by the following picture:
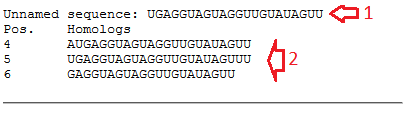
Figure 2. The output window.