MiRNA sites prediction
Program description: The tool searches for MiRNA sites by calculating miRNA binding efficiency.
Biological task that could be solved: Predicting the potential miRNA binding sites in an arbitrary target sequence.
Data input: Into the text-box (shown by the red arrow #1 in Fig.1), enter or insert from the clipboard a genomic sequence to be analyzed. You may input this sequence in the fasta format or in a plain text format without the comment line. The size limit is 10 000 symbols. Use the alphabets ATGC(atgc) or AUGC(augc), any other symbol in nucleotide will give an error. Line foldings and spaces are ignored.
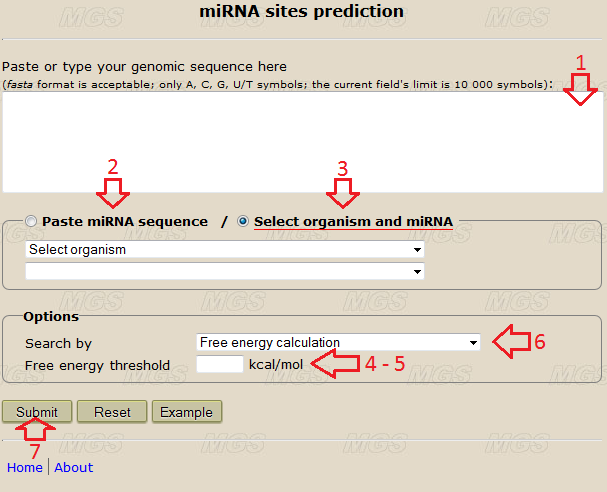
Figure 1. The main program window.
Program options: You may input one miRNA sequence in the text-field (red arrow #2), or select an organism and a desired miRNA (or all miRNAs) from drop-down lists (red arrow #3, you need to have JavaScript enabled in your browser, the miRNA sequences and the list of the organisms are from miRBase.org, release 17.0). For the "partition function" method you need to select only one miRNA sequence, for other methods you may also select all miRNAs from a particular organism. Use the alphabets ATGC(atgc) or AUGC(augc), any other symbol will give an error.
• "Free energy threshold" (red arrow #4) - this parameter specifies the largest free energy for a miRNA-RNA duplex in "free energy calculation" methods, it is required only for these methods. Default value (empty field) is 99999 kcal/mol (the calculation without an energy threshold). The value of this parameter is limited only by the size of the text-field. The more is "Free energy threshold" value, the less the prediction accuracy and the more number of sites will be predicted. Recommended value equals to -10,0.
• "Concentration parameter" (red arrow #5) - this parameter represents an apparent miRNA concentration on the target sequence in the "partition function" method, it is required only for this method. Default value (empty field) is 0, recommended value equals to 3. Minimal value equals to 0, maximal value is limited only by the size of the text-field. The more is the "concentration parameter" value, the less is the binding specificity calculated.
• Click the check-box "Search by" (red arrow #6) for choosing the method you want to apply:
- The "free energy calculation" method is based on the calculation of the miRNA-RNA duplex thermodynamic characteristics.
- The "5' context search" method assumes the full complementarity 2-8 positions of 5' miRNA ends and the target sites.
- The "free energy calculation and 5' context search" method combines both previous methods.
- The "partition function" method plots the graph of miRNA binding probability at each position of a target sequence.
Program execution: Start the program by clicking the button "Submit" (red arrow #7).
Data output: Program execution will bring up the resulting window with the data output. For the "free energy calculation" and "5' context search" methods the result includes the following (fig.2):
• The comment line from the text-box or the ID of the query miRNA and the name of the organism (if defined, red arrow #1);
• The sequence of the query miRNA (red arrow #2);
• The positions and the sequences of predicted sites grouped by the positions (red arrow #3).
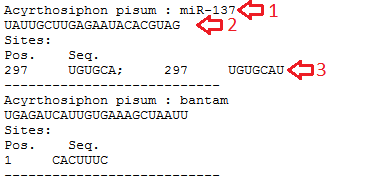
Figure 2. The resulting window for the "free energy calculation" and "5' context search" methods.
For the "partition function" method the program displays the graph of binding probability (fig.3) at each position of the target sequence except 20 positions in the beginning.
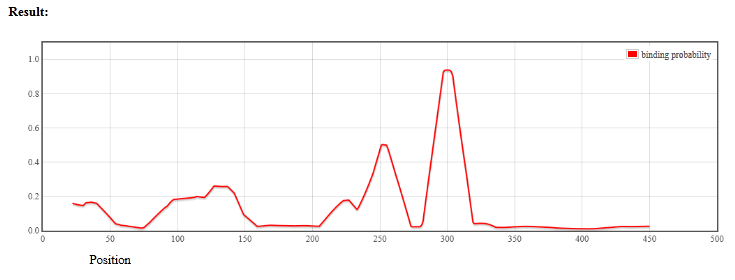
Figure 3. The resulting window for the "partition function" method.