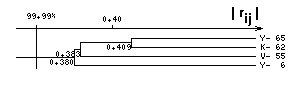![]()
![]()
![]()
![]()
![]()
![]()
|
|
|
|
CRASP: software package for analysis of physicochemical parameters of aligned sequences of protein families*Dmitry A. Afonnikov
Laboratory of Theoretical Genetics, Institute of Cytology and Genetics, Novosibirsk 630090, Russia; E-mail: ada@bionet.nsc.ru At present, the data on aligned primary sequences of protein families are being accumulated very rapidly. One of promising approaches to analysis of these data is to study correlations between amino acid substitutions at positions of protein sequences [1-3]. The information obtained could be valuable for revealing the peculiarities of the structure and function of the proteins under study. In relation to this topic, the topical goal is to develop publicly available methods and software for correlation analysis of protein sequences. In analysis of co-adaptive substitutions, two important tasks appear. The first is to study correlations of amino acid substitutions at positions of a protein in order to reveal the pairs of residues, which are related by functional interactions (e.g., steric contact). The resulted information could be used for prediction of possible contacts between the residues [4,5]. The second task is related to revealing and analysis of conserved integral physico-chemical characteristics of a protein [6,7]. The examples of these characteristics are the total charge of a protein molecule, the volume of its hydrophobic core, and hydrophobic moments of alpha helices, etc. The constancy of these characteristics in the course of evolution means that they are responsible for the key features in protein structure and function. Co-adaptive substitutions may serve as one of possible mechanisms for supporting the constancy of these characteristics, together with invariance of residues at positions of a protein and conserved substitutions of residues to similar by physico-chemical properties ones. In the present communication, we introduce the software package CRASP for analysis of physico-chemical pairwise correlations between amino acid substitutions at positions of aligned sequences of a protein family. In this package, we have realized an approach, which is built upon revealing conserved physico-chemical characteristics of a protein on the basis of information about correlations of substitutions of amino acids at pairwise positions of a protein. The software package is available via the Internet at http://wwwmgs.bionet.nsc.ru/mgs/programs/crasp/. By using the software package developed, it is possible
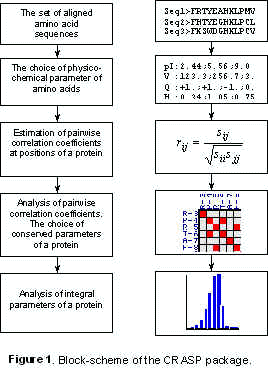
It should be noted that the software package consists of two modules: the program for analysis of correlations of substitutions at the pairs of protein positions and the program for analysis of integral protein characteristics. In principle, the user can address to each of these two blocks independently (in particular, if the physico-chemical characteristics is known by the user a priori). The block-scheme of the software package CRASP is shown in Fig. 1.
General methods and algorithmsThe basic methods for evaluation of dispersion, covariation between the values of physico-chemical characteristic of a protein, correlation coefficients between them, and estimation of significance level of dependencies revealed are described in our previous papers [8,9]. Below, we shall briefly describe the realization of these methods in the software package CRASP. Analysis of pairwise correlations of amino acid substitutions. To reveal correlations of amino acid substitutions at positions of a protein, an analysis is performed of the following protein physico-chemical characteristics: volume of a side group, charge, hydrofobicity, etc. These parameters for analysis are chosen by the user from the current version of the database on physico-chemical amino acid properties, which at present contains the set of 36 characteristics. It is supposed that these characteristics reflect the interactions between the residues in a protein globule. Hence, the revealed regularities between the values of physico-chemical characteristics of amino acids may indicate to existence of specific functionally important interactions between the residues. For analysis, the set of aligned amino acid sequences of a protein family is used. In analysis, each type of amino acid within alignment matrix is substituted to corresponding value of the chosen physico-chemical characteristics of amino acids. As the measure of relationships between amino acid substitutions at positions of a protein (columns of the resulted numerical matrix), we use the values of both linear correlation coefficient between the values of physico-chemical characteristics and partial correlation coefficients [8,9]. The partial correlation coefficients enable to evaluate the extent of direct relationships between the pair of protein positions, under condition that the residues in the rest positions stay invariant. Data weighting. To take into account the evolutionary relationships of the sequences analyzed, in the software package developed, it is possible to use various methods of data weighting. In the CRASP package, the possibility is realized to use the following data weighting schemes: weight calculation following the method by Vingron and Argos [10]; data weighting by accounting phylogenetic relationships in a protein family [9], or application of weight coefficients input by a user. A user produces the choice of the weighting method. Analysis of conserved integral characteristics of a protein. Integral characteristics of a protein F is determined as a linear combination of values of a certain physico-chemical property at positions of a protein [9]. For example, for the sequence with the index k, we get
where сi�s are some real numbers reflecting the impact of the residue at the i-th position into the value of integral characteristic Fk, L is the length of a sequence, fki is the value of physico-chemical property f for the residue at position i of the sequence k. As the measure of the constancy of the value F, for the set of sequences of a protein family, we use its sample dispersion D(F) [9]. In order to reveal conserved integral characteristics, we suggest to use information about pairwise correlations between the values of physico-chemical property at positions of alignment. Currently, the stage of revealing characteristics is not automated, so, a user makes the choice of integral characteristics personally. As the possible variants of such characteristics, we suggest to use the values of physico-chemical characteristic at the groups of such positions that have the maximal in absolute value correlation coefficients. To reveal such groups, an approach based on cluster analysis is realized in the package CRASP. For all positions analyzed, the procedure of clusterization is made on the base of the following measure of relatedness of the pair of positions i,j
that is, this distance is more the less, the most correlated are two positions of a protein. Clusterization is performed according to the nearest neighbor method. The results are given as a binary tree. Each node of this tree unifies two sets of positions (final vertexes of a tree that are the daughter vertexes for a given node); the location of a node in the dendrogram corresponds to the maximal correlation coefficient between all possible pairs compiled of positions from two different sets. In such a way, it is possible to reveal both pairs of correlating positions and the groups of positions, where substitutions are not accidental. To evaluate the extent of the constancy of the characteristics F, the value of its dispersion is compared to the value expected under assumption of independent substitutions at positions of a protein Dexp. This value may be estimated by the formula:
where rij are the linear correlation coefficients of a physico-chemical property at a pair of positions i,j (for independent amino acid substitutions, we set rij=0), and D(fi) is the value of a sampling dispersion of the value of physico-chemical property f at position i. In the software package CRASP, for evaluation of the F value constancy, the Monte-Carlo procedure is also used. Based on the model of evolution of physico-chemical protein characteristics, suggested earlier in [9], a large number of samples (up to 10000), with the size equaling to the size N of the set under analysis, is generated in the package CRASP. These samples are obtained by means of the Gaussian distribution of L independent variables, each is being characterized by the mean value and dispersion equal to their estimates for physico-chemical property of amino acids at positions of a protein. For each of these accidental samples, we estimate dispersions of a characteristic F, Drand(F) and Dexp(F), and calculate the value l = Drand(F)/Dexp(F). For the samples obtained, we evaluate the distribution of values Dexp(F), detected by the formula (1). While testing the hypothesis D(F) << Dexp(F), the share of the samples p such that Drand(F) > Dexp(F), is an estimate of the significance threshold of false positive estimates. Thus, 1-p is an estimation of the significance level of the constancy of physico-chemical character F. Let, for example, during the modeling of 1000 samples, there were found 10 out of them such that Drand(F) > Dexp(F). In this case, we consider that the constancy of the characteristic F is produced by co-adaptive substitutions of residues under the significance level equaling to 99%. Analogous estimations of significance level may be done for verifying the hypothesis D(F) >> Dexp(F). Analysis of J-domain �s sequencesBy means of the CRASP software package, we have analyzed a seria of protein families (CREB - AP-1 [9], homeodomains [14], experimental data on phage display [15,16]). In the work given, we have performed an analysis of multiple alignment of the J-domain sequences (extracted from the Pfam database, record PF00226 [11]). These domains are located in the N- terminal part of the DnaJ proteins, which are referred to chaperone system [12]. From the sample, the identical sequences and the sequences with a large number of deletions were eliminated, the threshold of variability at positions was equal to 7. The results of analysis have revealed that the group of positions corresponding to the numbers 6, 55, 62 and 65 demonstrate significant correlation coefficients between each other according to the value of isoelectric point of amino acids pI (Fig. 2а).
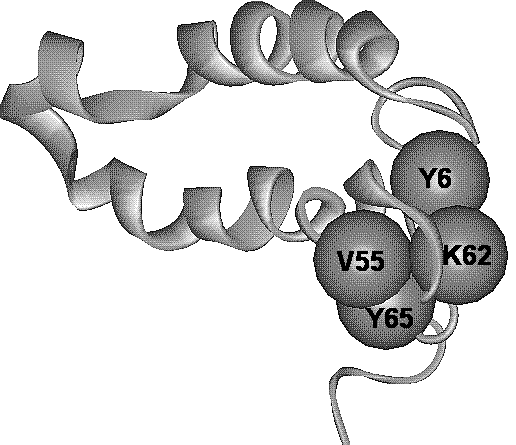
Notably, position 65 has positive correlation coefficients with the other positions from this group. On the contrary, positions 6, 62, and 55 have mutually negative correlation coefficients. Interestingly, within the spatial structure of DnaJ domain, the residues at these positions are closely located (Fig. 2b). We have supposed that due to compensatory effect (negative correlations), the total isoelectric point value of amino acids at positions 6, 62, and 55 (pI[6,62,55]) is conserved. On the other hand, the positive relationships of pI values at these positions with position 65 should support the constancy of the difference between the values pI[6,62,55] and pI[65]. Thus, we suggest that in general, for this group of positions, the co-adaptive substitutions of residues will support the constancy of the characteristic F=pI6+pI55+pI62-pI65. An analysis proved that dispersion of this value D(F) in the sample of DnaJ domains equals to 2.87, which is ~2 fold less than the value Dexp(F)= 5.73. Computational modeling gave the evidence that out of 10000 random sets, neither had the Drand(F) less than D(F). So, we may conclude that the constancy of the characreristics, which we have found, appears due to co-adaptive amino acid substitutions (at the error level p<10-4). It may be supposed that the constancy of this characteristic supports stability of mutual packaging of N- and C-terminal regions of the DnaJ domain. AcknowledgementsThe work is supported by the Russian Foundation for Basic Research (grants Nos 98-07-91078, 99-04-49879 and INTAS № INTAS-96-1787). The author is grateful to Galina Orlova for translation of the manuscript into English. References
|