Prediction of the DNA capacity to form nucleosome structure based on sequence statistics is of importance in analysis of gene expression regulation in eukaryotes. Context analysis of nucleotide sequences of experimentally defined nucleosome formation sites allows the determination of the sequence preference for nucleosome formation relying on statistical information. However, context analysis does not allow to identify the clear-cut consensus making feasible site prediction. One has to make recourse to more general context sequence features such as dinucleotide frequencies. Markov model is a common approach to the prediction of the functional regions in DNA sequences that disregards positional information. Here we use an improved version of the Markov model to predict the preference of DNA sequences to be within a nucleosome structure. The developed VMM program (the Variable Memory Markov model) computes the nucleosome formation potential for genomic DNA sequences of arbitrary lengths, including the short transcription factor binding sites.
The compaction degree of DNA within chromatin in the eukaryotic cells is different. The main structural element of chromatin, the nucleosome, is formed by a DNA fragment of about 147 bp packaged around a histone octamer (Khorasanizadeh, 2004). Experimentally defined binding sites of DNA to histone octamer underlie statistical analysis of nucleosome formation sites (Ioshikhes&Trifonov, 1993; See also latest Nucleosome database, Levitsky et al., 2005). The mechanisms of sequence-directed nucleosome positioning have been studied in numerous in vivo and in vitro experiments that suggested the existence of a specialized nucleosome code that determines this positioning as a result of multiple histone-DNA interactions (Kiyama&Trifonov, 2002).
The questions of the context specificity of nucleosome DNA in relation to the regulatory regions stir interest. It is feasible, in principle, to develop algorithms for the identification of nucleosome sites (Levitsky et al., 1999). Several approaches for nucleosome formation site analysis were suggested (Trifonov&Sussman, 1980; Ulyanov&Stormo, 1995; Stein&Bina, 1999). A periodic occurrence of certain dinucleotides rendering the bending ability of DNA double helix was shown by Trifonov&Sussman (Trifonov&Sussman, 1980). Difference of nucleosome formation sites from random sequence was demonstrated by Satchwell et al. (Satchwell et al., 1986) using Fourier analysis. Stein and Bina (Stein and Bina, 1999) show that the nucleotide triplet consensus non-T(A/T)G (abbreviated to VWG in 15-lettered code) influences nucleosome positioning and nucleosome alignment into regular arrays. This triplet consensus exhibits a fairly strong 10 bp periodicity in human DNA, implicating it in anisotropic DNA bendability. It is demonstrated that the experimentally determined preferences for nucleosome positioning in native SV40 chromatin can be predicted simply by counting the occurrences of the period-10 VWG consensus. Correlation with AA/TT periodicity also was shown (Ioshikhes et al., 1996). But no available software tool for such prediction was presented. Recently, Dalal et al. (2005) have experimentally examined the characteristics of nucleosome array formation in different regions of mouse liver chromatin, and have computationally analyzed the corresponding genomic DNA sequences. They provide evidence that computationally predictable information in the DNA sequence using VWG periodicity may affect nucleosome array formation.
Schematic presentation of location of two nucleosomes.
Software tools for estimation of the nucleosome formation potential (the tendency to bind histone octamer and to form nucleosome structure) of arbitrary nucleotide sequences have been developed in the RECON program by Levitsky et al., 2001.
The main results of prediction concern differences in nucleosomal organization of exon, introns and gene regulatory regions. Averaged profiles of nucleosome formation potential (fit function), calculated by VMM for set of exons and introns from EID database (Saxonov et al., 2000).
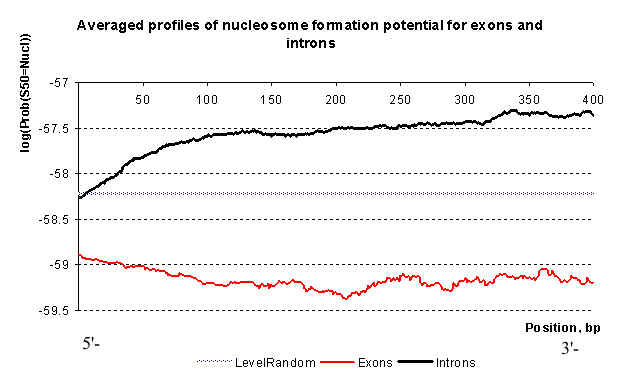
Averaged profiles of nucleosome formation potential for sets of exons and introns phased relative to 5'-end. Averaging was done for every position for all sequences in queried set. Downstream positions are not shown.
Features of nucleosome positioning code are imperfection and degeneracy. Therefore classical computer methods (alignment and consensus) are poorly applicable here. The RECON method is based on discriminant analysis and takes into account the frequencies of dinucleotides in the local regions of nucleosome sites. It is based on detection of the block structure of nucleosome formation site during its partition into local regions with a specific dinucleotide context.
The use of different approaches to the statistics based prediction of a sequence preference to be within a nucleosome structure broadens our benchtool for analysis of the genome structure and the regulatory mechanisms behind gene expression. Internet-available program VMM allows the calculation of the probability of a DNA region to be within a nucleosome structure.
A distinguishing feature of VMM is that it provides analysis of set of sequences and is capable of predicting the preference to be in a nucleosome structure for relatively short sequences of length less than 147 bp, including the transcription factor binding sites. The division into local regions used in RECON is inapplicable to analysis of short sequences; therefore, we use the more common Markov model. We implemented an expansion to the standard Markov model for the appearance of letters in a text, depending on the preceding context (Bejerano, 2004). The expansion we called the variable memory Markov model. This model uses more exhaustively than its predecessors the nucleotide correlations and it proved to be efficient in analysis of protein sequences.
Construction of such a Markov model is feasible for a given set of DNA sequences using the TreeComplexity program. The VMM program implements method for estimation of the fit of a DNA sequence to the predefined model of nucleosome formation site. see also Publications page.
A consensus sequence for nucleosome positioning has not been reported but certain DNA sequence preferences or motifs for nucleosome positioning have been discovered (Thastrom et al., 2004). To use all non-random motif we need mix several models based on such motifs. From the other side, model should be more or less homogenous like a Markov model, since there is no exact location of motifs in the nucleosome sites.
To estimate the fit of the queried sequence to the nucleosome site, we took advantage of the context tree source model. Otherwise defined, it is the variable memory Markov model. Like the standard Markov model, the newly developed is stationary, the probability of a nucleotide occurrence is not dependent on the position in a sequence. It is dependent on the local preceding (left) context only. The Xn = X1X2...Xn sequence is generated with probability
P(Xn) = P(X1|S1)P(X2|S2)…P(Xn|Sn).
Here, every S context is not longer a certain fixed length d that determines the probability distribution of a letter occurrence in a sequence immediately to the right. Contrary to the fixed order Markov models, these models are not restricted to a predefined order. Rather, by examining the training data, a model is constructed that fits higher order Markov dependencies where such contexts exist, while using lower order Markov dependencies elsewhere. As both theoretical and experimental results show, these models are capable of capturing rich signals from a modest amount of training data without using hidden states.
Ron et al (1996) have suggested the approach expressed as a subclass of probabilistic finite automata and they have demonstrated that an optimum model can be developed using the prediction suffix tree construction.
It is convenient to arrange all the possible contexts of fixed lengths into a tree-like structure.
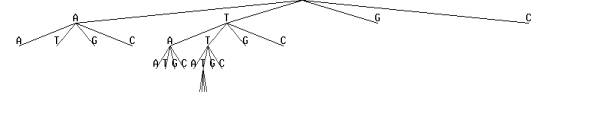
An example of context tree for nucleosome formation sites.
The probability P(Zi|Sj) of the
letter occurrence ZiÎ{A,T,G,C},
i=1,2,3,4 in every one of the Sj contexts is defined by the
respective q parameter as P(Zi|Sj)=q ij ,
where
![]() q
ij = 1,
j=1,2,...|T|, |T| is the number of all the contexts
in T model (the number of leaves in the tree), 4£|T|£4d,
d defines the maximum context length.
q
ij = 1,
j=1,2,...|T|, |T| is the number of all the contexts
in T model (the number of leaves in the tree), 4£|T|£4d,
d defines the maximum context length.
Contexts may vary in length, but no context is the end of some other context in the tree. Every path from leaves to root corresponds to the word in a DNA sequence. Every context has its own distribution for generation of the next letter in a sequence.
An optimum model of the generating tree source was built on the starting data using the Context algorithm (Barron et al., 1998). The algorithm for building the model is related to a mathematical estimation of stochastic complexity and the Internet-available program TreeComplexity implements it. Thus, not all the contexts present in the training sample are utilized.
The nucleosome formation potential was preformed by estimation of the correspondence probability of a sequence to the Markov model trained on the database. Fit function was constructed as the logarithm of the probability to obtain a sequence X in a sliding window of fixed length n:
F(Xn )
= log ( P(Xn) ) =
![]() log (P(Xn|Sj)
).
log (P(Xn|Sj)
).
The program outputs this profile together with the level expected by random for a DNA sequence with equal nucleotide frequencies. A window may be of any size, but it is expedient to have it smaller than 147 bp.
VMM provides the calculation of the fit function of a sequence not only to the nucleosome formation sites, but also to any DNA sequence set, whose tree model was constructed using the TreeComplexity program (http://wwwmgs.bionet.nsc.ru/mgs/programs/complexity/).
With the VMM program, users can analyze a single up to 1Mb sequence and obtain a profile of the preference for nucleosome formation. The program enables the calculation of the average profile for a set of phased (i.e. of the same length) DNA sequences. The latter possibility is of great interest for defining the preference for the formation of nucleosomes for different sequence sets, the gene promoters, for example.
With the given VMM method, to calculate the nucleosome potential, a homogenous model of a sequence is used. In contrast, the previous RECON method, the queried region of the nucleosome formation site was considered as a whole and the minimum length of the queried sequence was 160 bp. Thus, the model we propose allow the estimation of thee nucleosome potential for sequences as short as 20 bp. Comparison of nucleosome potential function based on variable memory Markov models and function based on dinucleotide frequencies and discriminant analysis revealed a correlation for nucleosome formation site sequences and genomic DNA. The proposed program is novel in that it allows the calculation of the nucleosome potential for short sequences, such as the transcription factor binding sites, the splicing sites.
An example of nucleosome prediction profile and experimental location of the nucleosomal site is presented in the picture.
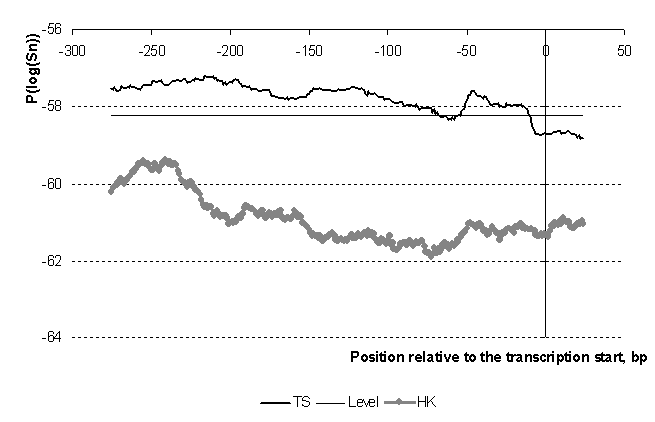
Nucleosome formation potential for eukaryotic gene promoters.
The specificity of the queried nucleosomal sites makes impossible accurate prediction for these sites. In fact, the nucleosome formation is determined not only by the DNA context features, but also by external influences, in particular the disposition of the neighbouring nucleosomes. The accuracy of most of the experimentally mapping data available in the literature is limited to 10-20 nucleotides or so. Even the very existence of a nucleosomal context code defined as degenerate periodic signals of particular nucleotides was debatable. Despite the questions raised, the program we propose nevertheless allows the delineation of regions where nucleosome formation is hindered. The important point is that only statistical data for the nucleosome formation sites from the NPRD database are used for the prediction.
The query of the sequence set allows to make statistical inferences about its average preference for nucleosome formation. We analyzed two sets of gene promoters phased relative to the transcription start as [-300; +100]. The samples were taken from the TRRD database (http://wwwmgs.bionet.nsc.ru/mgs/gnw/trrd) by the gene expression level: the promoters of housekeeping genes and the promoters of tissue specific genes. Figure presents averaged profiles for the samples.
The promoters of the housekeeping genes have smaller fit to nucleosome formation sites than promoters of tissue-specific genes. Thus, the results suggest that the genes expressed all the time in the nucleus should be devoid of tight chromatin packing unlike the tissue-specific genes whose expression is inducible by other factors. These results are in good agreement with our previous.
Indeed, there are some evidence that the basic repeating units of eukaryotic chromatin, nucleosomes, are depleted from active regulatory elements throughout genome in vivo (Lee et al., 2004). We found that during rapid mitotic growth, the level of nucleosome occupancy is inversely proportional to the transcriptional initiation rate at the promoter.
Taken together, the results showed that introns and 5'-untranslated gene regions have a smaller nucleosome formation potential than exons and gene regulatory regions (Supplementary Material 1).
The context features of the nucleotide sequence can be defined by various determinants that include protein coding, the RNA secondary structure and long-range correlations. These determinants define text complexity of a DNA sequence. Text complexity of long genomic DNA was estimated using the Complexity program. Nucleosome formation potential of the same sequences was estimated by the VMM program. As a result, a significant correlation of these parameters was established (Supplementary Material 2 and Supplementary Material 3). This supports and extents the idea that low text complexity of DNA sequences enhances nucleosome formation.
Analysis of text complexity of nucleosome formation site reveal symmetry and a M-letter distribution of averaged complexity profile.
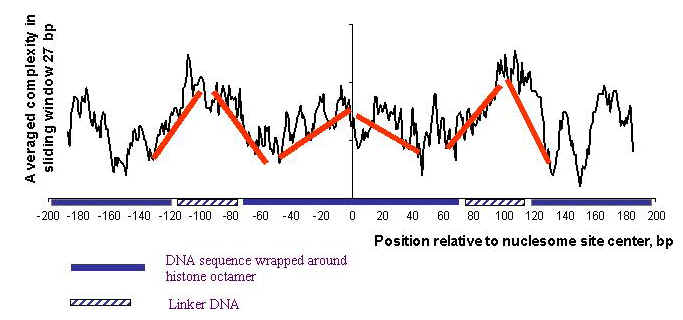
Averaged complexity of nucleosome formation sites (the modified Lempel-Ziv method)
Correlation of text complexity and nucleosome potential could be explained in terms of the superposition of many genetic codes: triplet code, code for RNA structure, code for protein secondary and tertiary structure and so on (Trifonov, 1997; Peleg et al., 2004).
Using the VMM program, we calculate the fit profile for sequences containing TFBS with flanks of length 100 bp. Every sample contained at least 40 sequences. We demonstrated that nucleotide sequences containing binding sites of HMG1, NFATp and Oct transcription factors have the greatest nucleosome formation potential (Supplementary Material 2). Indeed, the capacity of these protein transcription factors to bind DNA packaged in a nucleosome structure has been demonstrated.
DNA sequence is an important determinant of the positioning, stability, and activity of nucleosomes, yet the molecular basis of these effects remains elusive. A consensus DNA sequence for nucleosome positioning has not been reported and, while certain DNA sequence preferences or motifs for nucleosome positioning have been discovered, how they function is not known. Thastrom et al (2004) sugest that algorithms for aligning the selected DNA sequences should seek to optimize the alignment over much less than the full 147 bp of nucleosomal DNA. The alignment further reveals an inherent palindromic symmetry in the selected DNAs; it makes testable predictions of nucleosome positioning on the aligned sequences and for the creation of new positioning sequences.
Indeed, we observe symmetry in the source tree model obtained using nuclesome site and corresponding inverted sequences.
Recently, Vinogradov (2005) shown that the nucleosome formation potential of introns, intergenic spacers and exons of human genes is correlate with among-tissues breadth of gene expression. These results extent our promoter sequence analysis.
The VMM software allows the advantageous use of context properties of a DNA sequence promotes as a basis for independent predictions of nucleosome formation potential.
Bagga R., Michalowski S., Sabnis R., Griffith J.D. and
Emerson B.M. (2000) HMG I/Y regulates long-range enhancer-dependent
transcription on DNA and chromatin by changes in DNA topology. Nucleic Acids
Res., 28(13): 2541-50.
Barron A., Rissanen J. and Yu B. (1998) The minimum
description length principle in coding and modelling. IEEE Trans. Inform. Theory, 44: 2743-2760.
Belikov S., Holmqvist P.H., Astrand C. and Wrange, O.
(2004) Nuclear factor 1 and octamer transcription factor 1 binding preset the
chromatin structure of the mouse mammary tumor virus promoter for hormone
induction. J Biol Chem., 279(48): 49857-67.
Johnson B.V., Bert A.G.,
Ryan G.R., Condina A. and Cockerill, P.N. (2004) Granulocyte-macrophage
colony-stimulating factor enhancer activation requires cooperation between NFAT
and AP-1 elements and is associated with extensive nucleosome reorganization. Mol
Cell Biol., (18): 7914-30.
Kiyama R. and Trifonov
E.N. (2002) What positions nucleosomes? – A model. FEBS Lett. 523,
7–11.
Levitsky
V.G., Proscura A.P., Podkolodnaya O.A., Ignatieva E.V., Ananko E.A. (2004)
Nucleosome
formation potential of the gene regulatory regions.
Proceedings of the Fourth International Conference on
Bioinformatics of
Genome Regulation and
Structure (BGRS'2004), IC&G
Press, Novosibirsk, Vol.1,
p.130-133.
Pullner, A., Mautner, J., Albert, T. and Eick, D. (1996)
Nucleosomal structure of active and inactive c-myc genes. J Biol Chem., 271(49):
31452-7.
Ron D., Singer Y. and Tishby N. (1996) The power of
amnesia: learning probabilistic automata with variable memory length. Machine
Learning, 25, 117-149.
Additional materials including complete Proceedings of the BGRS conferences (2000, 2002 and 2004) in PDF format are accessible at the official Internet-site of the series of International Conferences on Bioinformatics of Genome Regulation and Structure.
![]()
This resource has been developed in Institute of Cytology
and Genetics, Novosibirsk, Russia
Authors: Yu.L.Orlov,
V.G.Levitsky
Team leader: N.A.Kolchanov
Version - 2005.