Ab initio human pre-miRNA prediction
Program description: The program for ab initio human pre-miRNA prediction in an arbitrary genomic sequence.
Biological task that could be solved: Identification of novel human pre-miRNAs.
Data input: Into the text-box (red arrow #1, Fig.1), enter or insert from the clipboard a genomic sequence to be analyzed. You may input this sequence in the fasta format or in a plain text format without the comment line. The size limit is 10 000 symbols. Use the alphabets ATGC(atgc) or AUGC(augc), any other symbol in the nucleotide sequence will give an error. Line foldings and spaces are ignored.
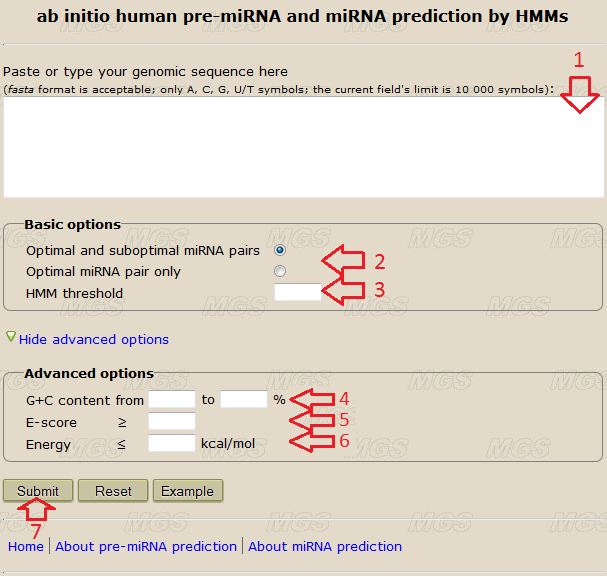
Figure 1. The main program window.
Program options: To classify a short (120nt or less in length) genomic sequence as a miRNA precursor you need to input it into the text-box (red arrow #1) and set the "HMM threshold" (red arrow #3). Leave the other fields (red arrows #4-6) empty. "HMM threshold" determines the largest possible HMM's score for the pre-miRNA candidates. When calculating without specifying the HMM threshold it’s default value is 1000. The higher is the "HMM threshold" value, the less is the prediction accuracy and the more number of pre-miRNAs are predicted (see additionally fig.3). Recommended value equals to 1.93.
For genomic sequences longer than 120nt the program takes more time to calculate. In this case we recommend to apply additional sequence filters and to adjust pre-miRNA prediction options (red arrows #4-6).
• "G+C content" (red arrow #4) - these parameters determine the biggest and the smallest values of G+C nucleotides percentage in a pre-miRNA candidate. Default values (empty fields) are 0% and 100%. The less is the difference between "G+C content" values, the faster is the calculation and less the accuracy. The values giving the accurate and at the same time fast prediction are 30% and 70%.
• "E-score" (red arrow #5) - this parameter sets the smallest value of E-score for a pre-miRNA candidate. It is calculated as (9*Ng*Nc + 3*Na*Nu + 2*Ng*Nu)/120, where Ng, Nc, Na, Nu - the number of G,C,A,U/T nucleotides in sequence. Minimal value equals to 0, default value (empty field) is 0. The higher is the "E-score", the faster is the calculation and the less the accuracy. Value giving the accurate and at the same time fast prediction is 30.
• "Energy" (red arrow #6) - this parameter determines the highest free energy of a pre-miRNA candidate structure. The energy values are calculated by the GArna software. The value of this parameter is limited only by the size of the text-field. Default value (empty field) is 99999 (without an energy threshold).
Program execution: Start the program by clicking the button "Submit" (red arrow #7).
Data output: Program execution will bring up the resulting window with the data output.
They include the following (fig.2):
• The number of the predicted pre-miRNA (red arrow #1);
• The start position of this pre-miRNA in the genomic sequence (red arrow #2);
• The pre-miRNA's stem-loop with marked miRNA/miRNA* duplex for the optimal miRNA/miRNA* duplex (red arrow #3) or for the best two variants (red arrows #3-4). The miRNAs are marked by the uppercase letters.
• The sequences of the predicted miRNAs.
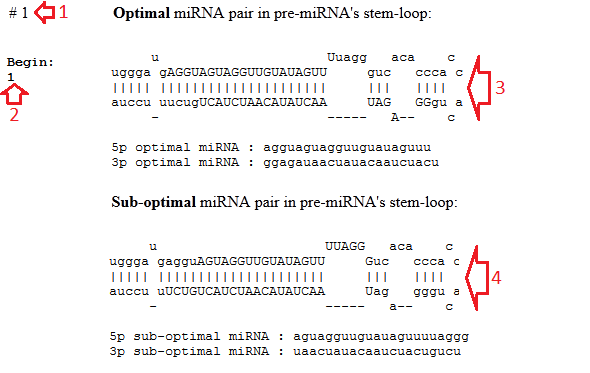
Figure 2. The resulting window.
Evaluating the performance of human pre-miRNA classification:
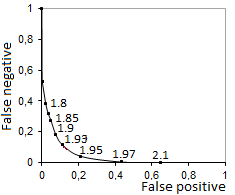
The text markers on the graph show the values of the HMM thresholds. The HMM was trained and validated through 5-fold cross-validation test with a positive dataset (known human pre-miRNAs, miRBase, rel.21.0) and a negative dataset (1881 random sequences from pseudo pre-miRMAs, proposed by Xue С. and co-authors]. Our method was tested without any constraints on the energy, G+C content and e-score.
Comparing the efficiency with other programs for pre-miRNA prediction:
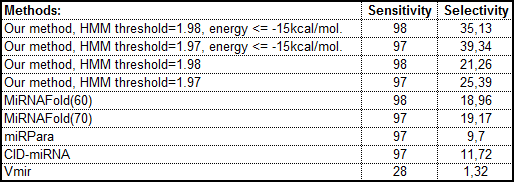
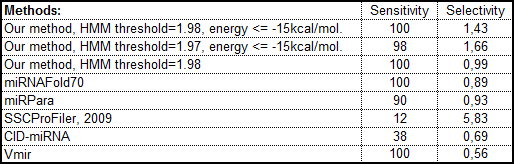
RERERENCES:
Xue С., Li А., He Е., Liu П., Li Y.,Zhang X. (2005) Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinformatics. 6, 310.
miRNAFold: Sebastien Tempel and Fariza Tahi. (2012) A fast ab-initio method for predicting miRNA precursors in genomes. NAR, 40(11): e80.
MiRPara: Wu,Y., Wei,B., Liu,H., Li,T. and Rayner,S. (2011) MiRPara: a SVM-based software tool for prediction of most probable microRNA coding regions in genome scale sequences. BMC Bioinformatics, 12, 107.
SSCProfiler: Oulas et al. (2009) Prediction of novel microRNA genes in cancer-associated genomic regions-a combined computational and experimental approach, NAR, 37(10): 3276-87.
CID-miRNA: Tyagi,S., Vaz,C., Gupta,V., Bhatia,R., Maheshwari,S., Srinivasan,A. and Bhattacharya,A. (2008) CID-miRNA: a web server for prediction of novel miRNA precursors in human genome. Biochem. Biophys. Res. Comm., 372, 831–834.
Vmir: Grundhoff,A., Sullivan,C.S. and Ganem,D. (2006) A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA, 12, 733–750.